





Abstract
Objective The mechanosensitive nonselective cation channel (NSCMS) and endothelin-1 (ET-1) play critical roles in the regulation of vascular tone. This study was undertaken to investigate the effect of ET-1 on NSCMS and on the myogenic response of arteries.
Methods Cell-attached patch-clamp techniques were applied to rabbit pulmonary and cerebral arterial smooth muscle cells using a 140 mM CsCl pipette and bath solutions (Ca2+-free, 1 mM EGTA). Myogenic responses were determined by video analysis of pressurized arteries.
Results The application of negative pressures through the pipette activated NSCMS, and this was augmented by bath application of ET-1 (1 pM–30 nM). ET-1 lowered the lowest pressure required for NSCMS activation. NSCMS facilitation by ET-1 was prevented by BQ-123 (1 μM, an ETA antagonist) but not by BQ-788 (1 μM, an ETB antagonist). Phorbol 12-myristate 13-acetate (PMA, 100 nM), a protein kinase C activator, also increased the activity of NSCMS. ET-1- or PMA-induced facilitation of NSCMS was abolished by GF109203X (10 μM), a protein kinase C inhibitor. Video analysis of pressurized cerebral artery showed inhibition of the myogenic response by the NSCMS channel blockers GsMTx-4 (5 μM) and DIDS (3–100 μM). Treatment with ET-1 (10 pM) augmented the myogenic response and this was inhibited by DIDS (30 μM).
Conclusion Stimulation of ET-1 receptor (ETA) facilitates NSCMS via a protein kinase C-dependent signaling pathway in rabbit arterial myocytes. Our findings suggest that NSCMS play a role in the myogenic response and its augmentation by ET-1.
This article is referred to in the Editorial by Coleman and Parkington (pages 197–198) in this issue.
1 Introduction
In many organs, blood flow is held at a relatively constant level when perfusion pressure changes substantially [1]. This autoregulation of blood flow is achieved in several ways including the myogenic contraction triggered by mechanical stretch of vascular smooth muscle [2]. Since vascular smooth muscle cells are depolarized as luminal pressure increases, the existence of mechanosensitive nonselective cation channels (NSCMS) provides a plausible explanation to the myogenic response. Actually, NSCMS have been found in a wide variety of cells including vascular myocytes [3–6]. Our previous studies showed that NSCMS are more abundant in pulmonary arterial smooth muscle cells (PASMCs) than systemic arterial myocytes of rabbits [7].
Endothelin-1 (ET-1) is a peptide hormone released from endothelium and vascular smooth muscle cells, and it is a highly potent vasoconstrictor [8,9]. ET-1 also induces mitogenic and vascular remodeling effects, which are critically required for several pathophysiological mechanisms [9–14]. The release and expression of ET-1 are triggered by various physicochemical stimuli, such as mechanical stress and hypoxia [15–19]. Moreover, it has been argued that ET-1 dependent signaling is critical for hypoxia-induced pulmonary vasoconstriction and chronic hypoxia-induced pulmonary hypertension [12,18,19]. In vascular myocytes, stimulation of the two known ET-1 receptors, ETA and ETB[20], increases cytosolic Ca2+ concentration ([Ca2+]c) via various ways which include phospholipase C (PLC)/InsP3-dependent release of Ca2+ from intracellular stores and the activation of Ca2+ permeable nonselective cation channels [20–26].
In addition to the direct vasoconstrictive effects, ET-1 also enhances myogenic vascular tone [10,13,27–29]. Because NSCMS may play a critical role in myogenic autoregulation, it might be expected that NSCMS are activated by ET-1. However, no previous study has investigated the effect of ET-1 on NSCMS. Moreover, it is unclear whether the augmented myogenic contraction by ET-1 is mediated by activation of NSCMS. Therefore, in the present study, we examined whether ET-1 modulates NSCMS in rabbit pulmonary and cerebral arterial myocytes. Also, we sought to identify the relevant intracellular signaling pathway and the relation between these actions of ET-1 on NSCMS and the myogenic response of arteries.
2 Materials and methods
2.1 Preparation of smooth muscle cells
New Zealand White rabbits (1.5–2.0 kg, n=75) of either sex were used for this study, and the investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Rabbits were fully anaesthetized with sodium pentobarbital (50 mg/kg) and simultaneously injected with heparin (100 U/kg). Pulmonary arteries (outer diameters between 0.5 and 1 mm), the main trunk of basilar arteries, and the second and third branches of middle cerebral arteries were used as sources of vascular smooth muscles. After arteries had been dissected and cut into small pieces, they were incubated in 1 mL of first digestion medium (Ca2+-free Tyrode's solution containing papain 1 mg/mL) for 10–15 min and in second digestion medium (Ca2+-free Tyrode's solution containing collagenase 2.8 mg/mL) for 10–20 min; both of the two digestion mediums contained serum albumin (1.5 mg/mL) and dithiothreitol (1.5 mg/mL). Cells were isolated from tissue strips by gentle agitation using a fire-polished glass pipette in high K+- and low Cl− storage medium. Isolated cells were maintained in the storage medium (see below) at 4 °C until required.
2.2 Experimental solutions
Ca2+-free normal Tyrode's solution contained (in mM): NaCl 143, KCl 5.4, MgCl2 1, glucose 5.5, and HEPES 5 adjusted to pH 7.4 with NaOH. High K+/low Cl− storage medium contained (in mM): KCl 50, l-glutamate 50, KH2PO4 20, taurine 20, MgCl2 3, glucose 20, HEPES 10, and EGTA 0.5, adjusted to pH 7.3 with KOH. The bath and pipette solutions for single-channel recording contained (in mM): CsCl 140, HEPES 10, EGTA 2 and MgCl2 1, adjusted to pH 7.4 with CsOH. Cells were immersed in CsCl bath solution for at least 30 min before performing electrophysiological recording. The bath used for patch recording was superfused by hydrostatic pressure (50 cm H2O) when a drug application was required. The bath solution for the pressurized artery system contained (in mM): NaCl 140, KCl 5.4, NaH2PO4 0.33, glucose 10, HEPES 10, CaCl2 1.8, and MgCl2 1 adjusted to pH 7.4 with NaOH.
2.3 Electrophysiological recording
Recordings were obtained in the cell-attach configurations using a patch-clamp amplifier (Axopatch 200A, Axon Instruments, Foster City, CA). Gigaseals were formed using Sylgard coated thin-walled borosilicate capillaries (Clark Electromedical Instruments, Pangbourne, UK). The negative pressure required to obtain gigaseals was applied minimally only during the initial phase of patch-clamp recording. Throughout the experiment, pipette pressure was equilibrated versus ambient pressure (i.e. 0 mm Hg). To activate NSCMS, negative pressure was applied through a patch pipette using a syringe connected to a water-filled meter [6,7]. Pressures (in cm H2O) were calibrated using a manometer and later converted into mm Hg. Electrical tip resistance of patch pipettes filled with experimental solution ranged between 2.3 and 2.5 MΩ. Care was taken to maintain a constant tip resistance since the size of the pipette tip affected cell membrane area and the degree of stretch induced by negative pressure. If not specifically mentioned, all NSCMS recordings were performed using a standard cell-attached patch-clamp setup (+50 mV, −15 mm Hg) at room temperature (22–24 °C). Polarities of clamp voltage refer to values on the cytoplasmic side. Recorded signals were filtered at 1 kHz and transferred to a computer using the Digidata 1200 interface (Axon Instruments, Foster City, CA) at a sampling rate of 5 kHz. Continuous single-channel currents were analyzed using the pCLAMP program (version 9, Axon Instruments). Channel activities are presented as NPo values, which was determined from continuous recordings of during ca. 1 min. Amplitudes of the single-channel currents at each membrane potential were obtained from all-point histograms.
2.4 Video analysis of pressurized arteries
Small arteries (250–350 μm, i.d., passive) was identified using a Nikon SMZ645 (Tokyo, Japan) stereo-zoom microscope. The dissected segments of small arteries were placed in a glass-bottomed vessel chamber (Model CH/1/SH, Living System Instrument, Burlington, VT, USA). The vessel chamber contained a HEPES-buffered physiological salt solution (pH 7.40±0.03; 140 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, 0.33 mmol/L NaH2PO4, 10 mmol/L glucose and 10 mmol/L HEPES), and its temperature was set at 37 °C by using a temperature controller (Model TC-01, Living Systems Instrument). Two pipettes were utilized to cannulate the vessel that was secured with 12-0 suture. To evaluate pressure-induced myogenic response, the luminal flow was maintained at zero. After confirming vessel integrity, intraluminal pressure was set to 40 mm Hg for at least an additional 60 min; spontaneous tone usually developed within this time frame. Vessel dimensions were measured using a video dimension analyzer (Model V94, Living Systems Instrument) and a data acquisition system (Digidata 1200 and Axoscope, Axon Instruments).
To assess myogenic behavior, internal diameters were measured during successive 20 mm Hg increments in luminal pressure over the range 20–100 mm Hg. After each step change in pressure, arteriolar diameter usually attained a steady state within 2 to 3 min. After a series of step changes, luminal pressure was returned to 40 mm Hg and the vessel concerned was allowed to re-equilibrate for a minimum of 5 min. At the end of each experiment, passive diameter measurements were recorded at each of the above pressures in 0 Ca2+ PSS containing 10 μM sodium nitroprusside. For pulmonary arteries, outer diameter was measured instead of inner diameter because the edge detector of our video analysis did not stably discriminate the margin of luminal wall of pulmonary arteries.
2.5 Drugs and chemicals
All drugs and chemicals except GsMTx-4 peptides were purchased from Sigma Chemical (St. Louis, MO). GsMTx-4 and its negative analogue were synthesized as reported previously [30].
2.6 Statistical analysis
Results are expressed as mean±S.E.M. Statistical significance was evaluated using the paired or unpaired Student t-test (P-value<0.05). N numbers in the Results section and in the figure legends indicate numbers of cells or vessels tested. The presence of NSCMS and their regulation by tested chemicals were consistently observed in this study, and were not confined to a particular animal. The numbers of rabbits sacrificed for each statistical summary of patch-clamp study were ten (Fig. 1), five (Fig. 2), seven (Fig. 3C,D), six (Fig. 3E,F), nine (Fig. 4C,D), seven (Fig. 4E,F), four (Fig. 7B, basilar artery), and three (Fig. 7B, cerebral artery), respectively.
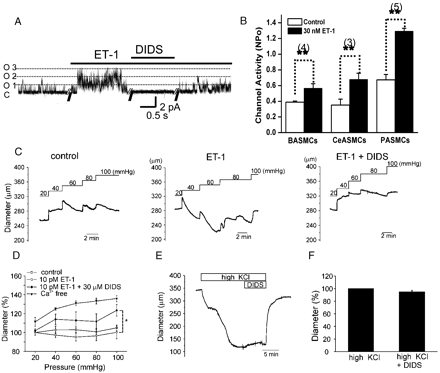
ET-1 induced facilitation of NSCMS and myogenic responses in rabbit cerebral artery. A. Representative current traces of cell-attached patches in middle cerebral arterial myocytes at a negative pressure of −15 mm Hg. Membrane voltage was clamped at 50 mV. The traces show that 30 nM ET-1 increased channel activity. Both basal and ET-1 induced activities were completely blocked by DIDS. B. The effects of 30 nM ET-1 on the NPo values of NSCMS in basilar, middle cerebral and pulmonary arterial myocytes. C. Representative traces of inner diameter from identical artery in control, pretreatment with 10 pM ET-1, combined treatment with ET-1 and 30 μM DIDS. The intravascular pressure was changed from 20 to 100 mm Hg as indicated. D. Summaries of the experiments shown in C including the full relaxed states in Ca2+-free condition (n=4). E. No effect of DIDS (30 μM) on the high K+ (60 mM)-induced constriction of cerebral artery. F. Summaries of the experiments shown in E (n=5). Diameters are expressed as a percent change of high K+-induced constriction.
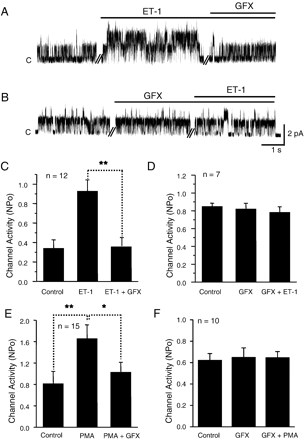
Inhibitory effects of GF109203X (GFX), a PKC inhibitor, on NSCMS facilitation by ET-1 and by phorbol 12-myristate 13-acetate (PMA). A, B. Representative current traces of cell-attached patches under continuous negative pressure of −15 mm Hg at 50 mV. Times of ET-1 (30 nM) and GFX (10 μM) application are indicated with bars above current traces. C, D. Summaries of the experiments shown in A and B, respectively. Numbers of tested cells are shown in the figure. E, F. Summaries of the effects of PMA and GF 109203X. GF 109203X was treated after (E) or before the application of PMA (F).
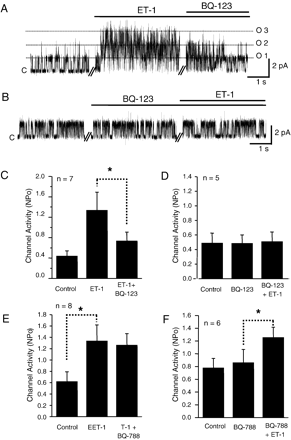
Blocking effects of ETA-selective antagonist (BQ-123) on ET-1-induced NSCMS facilitation. A, B. Representative current traces of cell-attached patches under a continuous negative pressure of −15 mm Hg at 50 mV. ET-1 (30 nM) and BQ-123 (1 μM) treatment times are indicated by bars above current traces. C, D. Summaries of the experiments shown in A and B. E, F. Unlike BQ-123, BQ-788 (1 μM, an ETB receptor antagonist) had no effect on NSCMS facilitation by ET-1. Numbers of tested cells are shown in each bar graph.
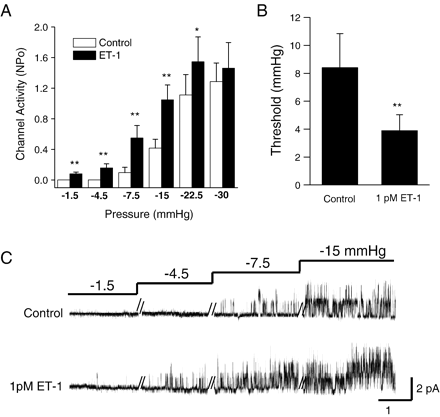
Increased NSCMS mechanosensitivity by ET-1. A. Summary of stretch-dependent increase in channel activity (NPo) with/without 30 nM ET-1 (n=8), **P<0.01; *P<0.05. B. Summary of the effects of 1 pM ET-1 on NSCMS activation pressure threshold (n=5). Negative pressure was gradually increased as shown in C to determine apparent threshold values. C. Original current traces showing NSCMS mechanosensitivity with/without 1 pM ET-1 in bath. The clamp voltage was 50 mV.
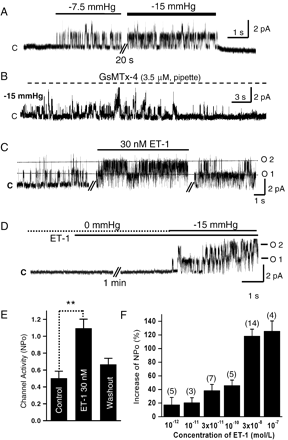
NSCMS inhibition by GsMTx-4 and their facilitation by ET-1 in rabbit PASMCs. Current traces recorded in cell-attached patches at +50 mV. The letters “c” and “o” represent closed and open channels, respectively. Numbers beside “o” represent open levels. A. Horizontal bars above the trace indicate the application of negative pressure (−7.5 mm Hg, −15 mm Hg) to the patch membrane. B. A representative trace recorded with 3.5 μM GsMTx-4 in the pipette solution. Channel activity was observed only during the initial membrane stretching period and then permanently disappeared while negative pressure (−15 mm Hg) was applied. C. Under the c–a condition, ET-1 increased NSCMS activity without activating other types of channels. D. The application of ET-1 without membrane stretch (0 mm Hg) did not activate NSCMS. Channel activity was evident only after applying negative pressure (−15 mm Hg). E. Statistical summary of the effect of 30 nM ET-1 on NSCMS activity (NPo) induced by membrane stretch (−15 mm Hg) in 14 PASMCs, **P-value<0.01. F. Summary of NPo changes induced by various concentrations of ET-1. Tested cell numbers are indicated above each bar. NPo values after ET-1 treatment were normalized versus control NPo in each membrane patch.
3 Results
3.1 Recording of NSCMS in arterial myocytes
Fig. 1 demonstrates representative current traces of NSCMS and response to ET-1 recorded in PASMCs. At a clamp voltage of 50 mV, negative pressure-induced single-channel currents with amplitude of about 1.5–1.8 pA, which corresponds to the known unitary conductance of NSCMS (30–33 pS) [16,17]. In the present study, the probability of recording NSCMS was about 50% in the PASMCs derived from the 3rd to 4th branch vessels (ϕ, 0.5–1.0 mm). In all patches confirmed to have NSCMS, no spontaneous activity was observed under control condition, i.e. without applying negative pressure. The effect of stretch was immediate and reversible: channel opening was turned on and off by applying and releasing pressure without a noticeable delay. As the magnitude of negative pressure increased, channel opening became more frequent and the number of open channels increased (Fig. 1A, see also Fig. 2).
NSCMS were inhibited by GsMTx-4, the venom of the tarantula Grammostola spatulata which possesses specific extracellular inhibitory effects on stretch-activated channels [31]. In c–a patches, GsMTx-4 was included in the pipette solution and stretch was applied as soon as the gigaseal has been formed. As demonstrated in Fig. 1B, when 3.5 μM GsMTX-4 was present in pipette solution, the initially observed stretch-activated channels disappeared permanently within 20–30 s after a gigaseal was formed. Similar results were obtained in three cases of c–a patch.
3.2 Effects of ET-1 on NSCMS
ET-1 (30 nM) increased NSCMS activity within 1–2 min of bath application, and this facilitation was maintained stably, and almost reversed by washout (Fig. 1C,E). In the absence of negative pressure, ET-1 did not activate any channels in the c–a condition even though NSCMS were present in the same membrane patch (Fig. 1D, n=33). Increases in NPo by 30 nM ET-1 at −15 mm Hg are summarized in Fig. 1E (n=14). NSCMS facilitation in the presence of negative pressure was observed from ET-1 concentrations of 1–100 pM (n=3–7 at each concentration, Fig. 1F). Although not shown, NSCMS facilitation by ET-1 was observed at both positive and negative membrane voltages and the slope conductance of NSCMS (33 pS) was not changed by ET-1.
The mechanosensitivities of NSCMS were compared in the presence and absence of ET-1 (Fig. 2A,B). Without ET-1, NSCMS activity was started to be observed from −7.5 or −15 mm Hg, but the application of 30 nM ET-1 sensitized NSCMS to lower negative pressures (−1.5 or −4.5 mm Hg). Interestingly, ET-1-induced NSCMS activity was less prominent at higher negative pressures (−22.5 and −30 mm Hg), which suggests that the effects of ET-1 are not simply additive. To elucidate whether sensitization of NSCMS sensitization to membrane stretching was also induced by lower concentrations, the apparent threshold pressure for activation was compared in the presence and absence of 1 pM ET-1. Interestingly, the lowest pressure required for NSCMS activation was found to decrease from 8.4±2.45 to 3.9±1.12 mm Hg (n=5) on adding 1 pM ET-1 (Fig. 2B,C).
Two types of ET-1 receptors, ETA and ETB, are specifically blocked by BQ-123 and BQ-788, respectively [9]. After confirming NSCMS facilitation by ET-1, we added 1 μM BQ-123 to bath solution, and found that NSCMS activity was reduced to the control level (Fig. 3A,C). Also, in the presence of BQ-123, ET-1 addition had no effect on NSCMS (Fig. 3B,D). In contrast, BQ-788 had no effect on ET-1 induced NSCMS facilitation (Fig. 3E,F).
It is known that ETA stimulation activates phospholipase C and protein kinase C (PKC) signaling cascades [9]. To test whether the effects of ET-1 are mediated via a PKC pathway, GF 109203X (10 μM, an inhibitor of PKC), was applied before or after the ET-1 treatment. As was observed for BQ-123, the application of GF 109203X (10 μM) in the presence of ET-1 returned NSCMS activity to the control level (Fig. 4A,C, n=4). Also, pretreatment with GF 109203X prevented NSCMS facilitation by ET-1 (Fig. 4B,D; n=4). Further supporting the involvement of PKC, the application of 100 nM phorbol 12-myristate 13-acetate (PMA, an activator of PKC) also facilitated NSCMS activity, and this activation by PMA was blocked by 10 μM GF 109203X (Supplementary Fig. 1). Responses to PMA and GF 109203X are summarized in Fig. 4E and F as bar graphs.
Because PLC activation via ET-1 receptors could also lead to stored Ca2+ release, we wondered whether the increased [Ca2+]c, in addition to PKC, might modulate NSCMS, or whether it might independently activate Ca2+-dependent nonselective cation channels. However, experimental conditions in the present study were designed to minimize the [Ca2+]c changes; myocytes were immersed in Ca2+-free CsCl solution (1 mM EGTA) for more than 20 min. Actually, when the [Ca2+]c of PASMCs under the above condition was measured by Fura-2 fluorometry, it was found that ET-1 (30 nM) did not increase [Ca2+]c (n=6, Fig. 5). In each case of this experiment, the large increase in [Ca2+]c by ionomycin in the presence of 10 mM CaCl2 was confirmed as a positive control.
![No effect of ET-1 on PASMC [Ca2+]c levels in Ca2+-free solution. In fura-2 loaded PASMCs, fluorescence ratio (340/380) was monitored. After a sustained incubation of PASMCs in Ca2+-free solution (1 mM EGTA, >20 min), the application of ET-1 (30 nM) had no effect on the fluorescence ratio. The Ca2+ sensitivity of cytosolic fura-2 was confirmed by adding ionomycin and 10 mM CaCl2.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/76/2/10.1016/j.cardiores.2007.06.021/2/m_76-2-224-fig5.gif?Expires=1716458843&Signature=ik3qpDPtSrOBafc3dDyZe6sIkZ3nbODkD8a60CLc4rR0M501pZvHeCSElg1Q8mvIXNn0zL4pPLbmQSg8eadasx~QnjiPxwcoaiOm47y1hSezB3QOLOK9PmsC3IDyS1QP2Rank4aZhADm36NFpYzD~LOveiXLA1UTnrlPfvTGY5dT5x3TZcqrHr7IwvK3zBum6IWsOHjRWpxUjd9G1qYNAb-S96PkfMAOt52QZcZqMrTqBmgFr9PG~-sHzXM9D0Edqqk9RCQAxHUOIzJUnV2s-hIRUkBUO3JxB5wmPYh~u0xAwuvhArS5TikOOpKtLVRryU5eA9mkHUzNs4AdQrqAkA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
No effect of ET-1 on PASMC [Ca2+]c levels in Ca2+-free solution. In fura-2 loaded PASMCs, fluorescence ratio (340/380) was monitored. After a sustained incubation of PASMCs in Ca2+-free solution (1 mM EGTA, >20 min), the application of ET-1 (30 nM) had no effect on the fluorescence ratio. The Ca2+ sensitivity of cytosolic fura-2 was confirmed by adding ionomycin and 10 mM CaCl2.
3.3 Myogenic response and NSCMS
To investigate the role of NSCMS and their modulation by ET-1 in terms of myogenic response in blood vessels, the change of inner diameter was measured in pressurized cerebral arteries. Arterial diameters were either unchanged or reduced in response to luminal pressure increases, indicating the presence of myogenic constriction. The application of GsMTx4 (5 μM) attenuated, but did not completely abolish this response in cerebral arteries (n=4, Fig. 6A,C). Our previous study [6] and the result shown below (Fig. 7A) show that DIDS blocks NSCMS. Consistent with the suspected role of NSCMS, the myogenic response was also inhibited by DIDS (Fig. 6B,D). Interestingly, the inhibitions by GsMTx-4 and DIDS were more prominent at the relatively low pressures, and the slow development of myogenic response was less affected by these blockers. When various concentrations were tested, DIDS showed more potent inhibitory effects on myogenic responses at lower luminal pressure range (Fig. 6E). Due to the high cost of experimentation with GsMTx-4, DIDS was used as a NSCMS blocker in the following experiments.
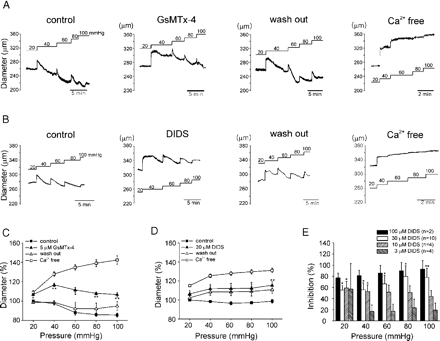
Myogenic responses of rabbit cerebral artery and effects of NSCMS blockers. A, B. Representative traces of inner diameter measured in isolated middle cerebral arteries. Arteries were subjected to step-wise increases in luminal pressure from 20 to 100 mm Hg, as indicated. Each panel indicates control, pretreatment of either 5 μM GsMTx-4 (A) or 30 μM DIDS (B), after washout of the drugs, and fully relaxed with Ca2+-free solution containing 1 mM EGTA and 10 μM SNP (Ca2+free). C. Summary of the effect of 5 μM GsMTx-4 on myogenic responses (n=4). Measurements of inner diameter were taken at steady state of each pressure-step. Diameters are expressed as percent values versus the control diameter at 20 mm Hg. *P-value<0.05, **P-value<0.01. D. Summary of the effect of 30 μM DIDS on myogenic responses (n=10). E. Summary of the inhibitions in myogenic tone induced by various concentrations of DIDS (n=2–10). Myogenic tones were initially measured as a percent decrease of the fully dilated diameter (obtained in 0 Ca2+ PSS+1 mM EGTA+10 μM SNP) of individual arteries at a given intravascular pressure. Then, the inhibitory effects of DIDS were expressed as a percent decrease of myogenic tone on a given pressure.
The effect of ET-1 on NSCMS was also examined in basilar and middle cerebral arterial myocytes (Fig. 7A,B). The blocking effects of DIDS on NSCMS in cerebral arterial myocytes were also confirmed (n=3, Fig. 7A). To compare responses with those of pulmonary artery, PASMCs were isolated on the same date and the effect of ET-1 was examined. As reported previously [7], control NSCMS activity was higher in PASMCs than in basilar and cerebral arteries (Fig. 7B). However, ET-1 (30 nM) also increased the activity of NSCMS in basilar artery (n=4) and middle cerebral arteries (n=3), and this effect was reduced by PKC inhibitor (data not shown here, n=2).
The application of 10 pM ET-1 did not significantly reduce the diameter of middle cerebral artery at 20 mm Hg but augmented myogenic constriction at higher luminal pressures (Fig. 7C), and the subsequent addition of DIDS reversed this augmented myogenic response (Fig. 7C,D). In contrast with its effects on myogenic responses, treatment with 30 μM DIDS did not inhibit vasoconstriction induced by high KCl (60 mM, Fig. 7E,F). The lack of an effect of DIDS on KCl-induced constriction indicates that it does not directly inhibit L-type Ca2+ channels or contractile molecules in vascular smooth muscle.
Since the distribution of NSCMS and their mechanosensitivities are higher in PASMCs than in systemic arteries, we also investigated pulmonary artery myogenic response. Unfortunately, unlike that observed for cerebral arteries, typical myogenic responses were not observed at luminal pressures between 5 and 25 mm Hg (physiological pulmonary arterial pressures). In fact, a slow decrease in diameter in response to raised luminal pressure was observed in only two of 25 trials (Supplementary Fig. 2). We also tested whether repetitive increases in luminal pressure from 5 to 15 mm Hg induced myogenic responses. In four of six pulmonary arteries, the application of ET-1 (100 pM) decreased inner diameters at 5 and 15 mm Hg, and this was subsequently reversed by DIDS (30 μM, Fig. 8). The other two arteries showed only passive dilation either in the absence or presence of ET-1 or DIDS (data not shown).
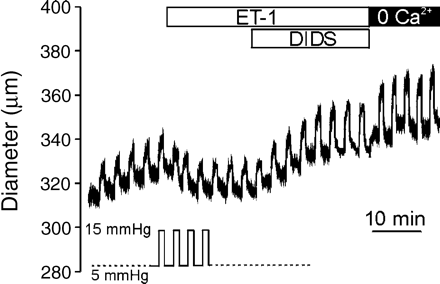
Changes of pulmonary arterial diameter in response to intravascular pressure. Trace of outer diameter in response to repetitive increase of intravascular pressure from 5 to 15 mm Hg for 1 min with 2 min of interval (see protocol in the figure). The sequence of applying ET-1 (100 pM), DIDS (30 μM) and washout with Ca2+ free solution (0 Ca2+) is indicated with bars above.
4 Discussion
The present study demonstrates that the mechanosensitivity of NSCMS in rabbit arterial myocytes is increased by ET-1 via ETA receptors in a manner that involves the PKC-dependent signaling pathway. The pressure–activity relation of NSCMS was shifted to the left by ET-1 (Fig. 2A). Also, cerebral artery myogenic tone was augmented by ET-1 and NSCMS blockers attenuated myogenic tone under control conditions and that augmented by ET-1. Previous studies have shown that ET-1 modulates various kinds of ion channels, e.g. it inhibits KATP and Kv channels [32–34], and stimulates L-type Ca2+ channels, Cl− channels, and nonselective cation channels [23,35–39]. These changes commonly imply that ET-1 has a depolarizing effect on vascular smooth muscle cells. Moreover, the facilitation of NSCMS by ET-1 would also be expected to depolarize arterial myocytes especially in vivo where cells are exposed to continuous mechanical stimuli.
Because stimulation with ET-1 is known to activate nonselective cation channels in vascular myocytes via signaling cascades like phospholipase C [37–39], we were initially concerned as to whether increased channel activity reflects NSCMS facilitation or an additive activation of receptor-operated cation channels. However, the present study demonstrates that the application of ET-1 alone does not activate single-channel currents (Fig. 1D). In contrast, once the presence of NSCMS has been confirmed in test membrane patches, channel activity was always increased by ET-1 (n=33). Therefore, we believe that the present study demonstrates genuine facilitation of NSCMS by ET-1. Nonetheless, considering previous report on ET-1 activated cationic currents in whole-cell recordings [37–39], the absence of channel activation by ET-1 alone in the present study is difficult to explain. The Ca2+-free condition might be supposed as a factor suppressing the activation of other cation channels by ET-1.
The effects of BQ-123 and of PKC agonist and antagonist on the modulation of NSCMS by ET-1 indicate that ETA receptors mediate NSCMS facilitation via a PKC-dependent signaling pathway. Similarly, ATP-sensitive K+ channels are also inhibited by ETA via PKC-dependent signaling in rabbit PASMCs and coronary arterial myocytes [33] whereas PLC-InsP3 dependent Ca2+ release is mainly mediated by ETB. The expression of both ETA and ETB in the rabbit PASMCs has been confirmed by an immunoblot assay [26]. Therefore, ET-1 receptor subtypes appear to regulate different targets in vascular myocytes.
Although the present study demonstrates PKC involvement in NSCMS facilitation by ET-1, it does not determine how PKC-dependent signaling, i.e. protein phosphorylation, actually changes NSCMS mechanosensitivity. Since the molecular nature of NSCMS in vascular myocytes remains controversial, we were unable to target a specific site for PKC phosphorylation. TRPM4 was recently proposed to be a mechanosensitive cation channel protein responsible for the myogenic response, and is activated by PKC [40]. TRPM4 was originally considered as a molecular candidate of Ca2+-activated nonselective cation channel, and the PKC-dependent phosphorylation of TRPM4 was reported to sensitize it to intracellular Ca2+[41]. However, in the present study, NSCMS were well-demonstrated under Ca2+-free conditions, which is inconsistent with the key property of TRPM4 as a Ca2+-activated channel [40,41]. TRPC6 has also been proposed as a NSCMS in vascular myocytes, and is known to be positively modulated by diacylglycerol [42], similar with the property of NSCMS reported by us [6]. However, the activation of PKC does not stimulate but rather desensitizes TRPC6 [43]. Therefore, none of the currently proposed candidates well-match the properties of NSCMS, and the precise mechanism of mechanosensitive activation and its facilitation by PKC remain to be determined.
NSCMS are regarded to be probably involved in the mechanism underlying arterial myogenic tone. In this respect, NSCMS facilitation might have implications in terms of increased myogenic response by ET-1 [10,13,28]. Since the inner diameter of the patch pipette was around 1 μm, when stretched by −15 mm Hg of pressure, a hemispheric plasma membrane in the pipette tip would be under the tension of 20 dyn/cm or higher depending on the geometry of a patch membrane. The reported wall tension of arteries for the study of myogenic response is around several hundreds of dyn/cm [44], and this seems to be sufficient to activate NSCMS. However, in the intact vessel, the wall tension is more likely to be transmitted by extracellular matrix and intracellular cytoskeletons rather than by direct stretch of plasma membranes. Therefore, one should be cautious to correlate the results of isolated myocytes with intact vessel study.
The present study demonstrates that myogenic responses shown by rabbit cerebral arteries and their augmentation by ET-1 are suppressed by NSCMS blockers, although the drug effects were incomplete. Interestingly, the myogenic responses were found to be generally more sensitive to NSCMS blockers at relatively low pressure ranges (Fig. 6). These results suggest that mechanism(s) other than the NSCMS could be also involved in the myogenic responses, especially at higher luminal pressure. Further investigation is required to determine the NSCMS-independent component of myogenic responses.
In contrast with the clear demonstration of myogenic responses in cerebral arteries, changes in luminal pressure with physiological range rarely induced myogenic response-like diameter decrease in pulmonary arteries. In fact, the presence of myogenic response in pulmonary arteries from normal adult animal is controversial [45–47]. In this respect, it is not likely that the NSCMS are active under the low luminal pressures of pulmonary circulation. However, the inhibition of ET-1 induced contraction by DIDS suggests that the facilitated NSCMS might participate, at least to a partial extent, in the agonist-induced vascular tone (Fig. 8).
Although typical myogenic responses are difficult to show, the majority of patch-clamp experiments have been performed in PASMCs because they have higher NSCMS densities and mechanosensitivities than systemic arterial myocytes [7]. In addition to this practical advantage, ET-1 is known to play a crucial role in the pathophysiology of the pulmonary circulation [11,12,48]. In view of ET-1 release under hypoxic conditions, our study suggests that NSCMS might participate in the hypoxia-induced changes of pulmonary arteries. The pulmonary arteries from pulmonary hypertensive animals or under chronic hypoxia started to show myogenic responses and altered ion channel expression [12,48,49]. In this respect, further investigations using specific pharmacological tools and the elucidation of the molecular identity of NSCMS are required to determine the roles of NSCMS in the pulmonary circulation and in classical myogenic responses of systemic arteries.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.cardiores.2007.06.021.
Acknowledgement
This work was supported by the Korea Research Foundation funded by the Korean Government (MOEHRD, Basic Research Promotion Fund, KRF-2006-E00021).
References
Author notes
These authors contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}