





Abstract
Pulmonary hypertension is a progressive disease with poor prognosis, characterized by pathological inward remodelling and loss of patency of the lung vasculature. The right ventricle is co-affected by pulmonary hypertension, which triggers events such as hypoxia and/or increased mechanical load. Initially the right ventricle responds with ‘adaptive’ hypertrophy, which is often rapidly followed by ‘maladaptive’ changes leading to right heart decompensation and failure, which is the ultimate cause of death.
We report here that miR-223 is expressed in the murine lung and right ventricle at higher levels than in the left ventricle. Moreover, lung and right-ventricular miR-223 levels were markedly down-regulated by hypoxia. Correspondingly, increasing right-ventricular load by pulmonary artery banding, induced right-ventricular ischaemia, and the down-regulation of miR-223. Lung and right ventricle miR-223 down-regulation were linked with increased expression of the miR-223 target; insulin-like growth factor-I receptor (IGF-IR) and IGF-I downstream signalling. Similarly, miR-223 was decreased and IGF-IR increased in human pulmonary hypertension. Notably in young mice, miR-223 overexpression, the genetic inactivation or pharmacological inhibition of IGF-IR, all attenuated right-ventricular hypertrophy and improved right heart function under conditions of hypoxia or increased afterload.
These findings highlight the early role of pulmonary and right-ventricular miR-223 and the IGF-IR in the right heart failure programme initiated by pulmonary hypoxia and increased mechanical load and may lead to the development of novel therapeutic strategies that target the development of PH and right heart failure.
1. Introduction
Pulmonary hypertension (PH) is a progressive disease of multifactorial aetiology, which has a poor prognosis and results in right heart failure. The vascular pathology of PH is characterized by pulmonary vasoconstriction, and by abnormal inward remodelling processes that result in severe loss of cross-sectional area and a concomitant increase in right-ventricular afterload. 1 , 2 Such structural changes suggest a switch from ‘quiescent’ towards ‘pro-proliferative’, ‘apoptosis-resistant’, and ‘pro-inflammatory’ vascular cell phenotypes. As a functional consequence, the pulmonary vascular resistance markedly increases, causing enhanced afterload for the right ventricle (RV) with right heart hypertrophy and failure as further cardiac sequelae of the disease. 2–4 Indeed, right heart failure is the cause of death of most patients with severe PH.
Deciphering the molecular mechanisms that drive the maladaptive inward remodelling processes in PH is indispensable for the development of therapeutic approaches to prevent or reverse such processes as is the identification of pathways activated in parallel in the lung and right heart. The latter requires better understanding of the switch from ‘quiescent’ to ‘proliferative’ cell phenotypes, disruption of the vicious pathogenic circuits that drive angio-proliferative abnormalities, and activation of repair and regenerative mechanisms. Extrapolating molecular mechanisms involved in the development of failure from the left to the right heart is, however, complicated by the fact that the two chambers have distinctly different embryological origins, the major differences in architecture, basic physiology, and the extent of the increase in afterload under hypertensive conditions. 5 , 6
Many of the changes in gene expression (e.g. inflammatory cytokines, growth factors as well as matrix remodelling proteins and matrix metalloproteinases) that contribute to the process of remodelling are regulated by hypoxia-induced transcription factors; in particular hypoxia-inducible factor (HIF)-1α. More recently, it has become clear that additional regulatory mechanisms may be involved in the response to hypoxia, particularly as low oxygen tension has been reported to determine the expression of several microRNAs (miRNAs), 7 , 8 which in turn can regulate angiogenesis and remodelling. 9 , 10 While several miRNAs have been linked with PH, 9 , 11–22 relatively little is known about the miRNAs regulated by PH in the pulmonary artery—right heart axis, their target proteins, and biological impact. Therefore, the aim of the present investigation was to determine which miRNAs are altered in chronic hypoxia-induced PH and right heart hypertrophy and to determine their role, if any, in hypoxia-induced right heart dysfunction.
2. Methods
2.1 Animals
C57BL/6 mice (6–8 weeks old) were purchased from Charles River (Sulzfeld, Germany). miR-223 knockout mice (miR223 y/− ) 23 were provided by Fernando D. Camargo (Harvard University, Cambridge, MA, USA), and bred by the animal facility at the University of Frankfurt. As the miR-223 locus is located on the X chromosome, miR-223 −/y mice and their miR-223 +/y littermates were studied. HIF-1α oxygen-dependent degradation domain (ODD)-luciferase mice (FVB.129S6-Gt(ROSA)26Sortm1(HIF1A/luc)Kael/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice with the tamoxifen-inducible deletion of the IGF-IR in cardiac myocytes (iCMIGF-IRKO mice) were generated as described. 24 Cre negative and Cre positive littermates were injected with tamoxifen (100 µL i.p. of 20 mg/mL in peanut oil) every day for 10 days and experiments started 10 days after the last tamoxifen injection. Mice were housed in conditions that conform to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH publication no. 85–23). Both the University Animal Care Committees and the Federal Authorities for Animal Research at the Regierungspräsidium Darmstadt (Hessen, Germany; #F28–21; #F28–47) and Regierungspräsidium Giessen (Hessen, Germany; #GI20/10 Nr. 103/2010 and Nr. 6/2012) approved the study protocol.
2.2 Hypoxia
Mice were housed in a ventilated chamber (BioSpherix, Lacona, USA) for up to 21 days and maintained under either normoxic conditions (21% O 2 ) or exposed to normobaric hypoxia (10% O 2 ). Some of the C57BL/6 mice exposed to normoxia or hypoxia were also treated with either PBS, control antagomir (GFP specific), antagomir directed against miR-223 (both 8 mg/kg, 100 µL; VBC Biotech, Vienna, Austria) twice per week for 21 days, or with adeno-associated virus (AAV) vectors for the cardiac-specific overexpression of pre-miR-223 or EGFP as a control. The cardiac-specific AAV vectors were generated as described. 25 , 26 Another group of animals were treated with GSK1904529A (30 mg/kg p.o.), a small-molecule inhibitor of the IGFR tyrosine kinase (Selleck Chemical LLC, USA). 27 Treatment with the inhibitor or its corresponding vehicle, β-cyclodextrin sulfobutylether (CyDex Pharmaceuticals Inc., USA) began on Day 8 after placing mice in the hypoxia chamber and was given daily for 2 weeks. Thereafter, haemodynamic parameters and RV hypertrophy were assessed under isoflurane anaesthesia (initially 3%, thereafter 1.5–2.0%) administered via a non-invasive nose cone as described, 28 or animals were sacrificed by exsanguination and organs were removed for further analysis.
2.3 Pulmonary artery banding
Adult male mice (Charles River Laboratories, Sulzfeld, Germany) weighing 20–23 g at the time of surgery were subjected to banding of the main pulmonary artery or sham operation under isoflurane anaesthesia (1.5% vol/vol) and a subcutaneous administration of 0.03 mg/kg buprenorphine hydrochloride, as described. 29
2.4 MicroRNA array
Total RNA was isolated from lungs of mice (four animals per group) kept for 3 weeks under normoxic (21% O 2 ) or normobaric hypoxic (10% O 2 ) conditions. The array analysis (360 mouse microRNA genes) was performed by DNA VISION (Gosselies, Belgium). Final data were presented as relative expression of miRNAs in hypoxia vs. normoxia.
2.5 Human material
The study protocol for tissue donation was approved by the Ethics Committee (Ethik Kommission am Fachbereich Humanmedizin der Justus Liebig Universität Giessen) of the University Hospital Giessen (Giessen, Germany) in accordance with national law and with ‘Good Clinical Practice/International Conference on Harmonisation’ guidelines and the Declaration of Helsinki. Written informed consent was obtained from each individual patient or the patient's next of kin (AZ 31/93).
2.6 Statistics
Data are expressed as the mean ± SEM, and statistical evaluation was performed using Student's t -test (two-tailed) for analysis between two groups containing normally distributed data and one- or two-way ANOVA followed by Newman–Keuls multiple comparison test, values of P <0.05 were considered statistically significant.
For detailed methods see Supplementary material online .
3. Results
3.1 Chronic hypoxia regulates miR-223 expression in the mouse lung and right heart
In mice maintained under hypoxic (10% O 2 ) vs. normoxic (21% O 2 ) conditions for 21 days, 26 miRNAs were altered by more than 30% ( Supplementary material online, Table S1 ). The miRNA increased to the greatest extent by hypoxia was miR-210; referred to as the master hypoxamir, 30 , 31 while the most down-regulated (∼50%) highly expressed miRNA was miR-223 ( Figure 1 A ). In the lung, miR-223 expression was most prominent in vascular smooth muscle cells ( Figure 1 B ) and RT–qPCR and a miR-223 core promoter assay confirmed that hypoxia decreased miR-223 levels in vivo ( Figure 1 C ) and in vitro ( Figure 1 D and E ).
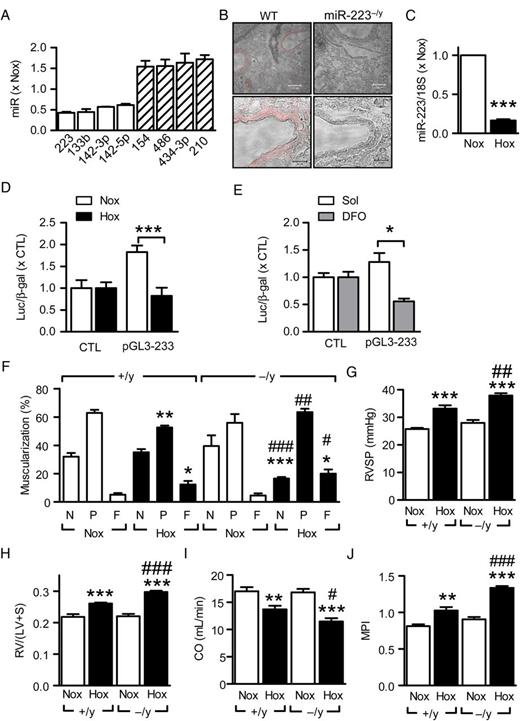
Hypoxia-induced regulation of miR-223 in the mouse lung. ( A ) MicroRNAs in murine lung most significantly altered by hypoxia (10% O 2 , 21 days) vs. normoxia (Nox); n = 4 per group. ( B ) Representative in situ hybridizations showing miR-223 (red) expression in wild-type (WT, +/y) and miR-223 −/y (−/y) lungs; bar = 100 µm (upper panels) and 20 µm (lower panels). ( C ) RT–qPCR analysis of miR-223 expression in normoxic and hypoxic (Hox) mouse lungs; three independent experiments with at least four mice per group and experiment; *** P <0.001, t -test. ( D ) Effect of normoxia (Nox) vs. hypoxia (Hox, 1% O 2 ) on miR-223 promoter activity in COS-7 cells; n = 4. ( E ) Effect of chemical hypoxia (i.e. deferoxamine; DFO; 130 µmol/L, 48 h) on miR-223 core promoter activity in COS-7 cells; n = 4. ( F ) Effect of hypoxia (Hox) on pulmonary artery (20–70 µm) remodelling in mouse lungs from +/y and −/y littermates, N = non-muscularized; P = partially muscularized; F = fully muscularized ( n = 5–8). ( G ) Effect of hypoxia on right-ventricular systolic pressure (RVSP) in +/y and −/y littermates ( n = 5–14). ( H ) Effect of hypoxia on right-ventricular hypertrophy (RV/(LV+S)) in +/y and −/y littermates ( n = 5–14). ( I and J ) Effect of hypoxia on right-ventricular function in wild-type (+/y) and miR-223 −/y littermates ( n = 7–9); ( I ) cardiac output (CO, mL/min) and ( J ) myocardial performance index (MPI). * P <0.05, ** P < 0.01, *** P <0.001 vs. Nox; #P <0.05, ##P <0.01, ###P <0.001 vs. +/y, ANOVA (Newman–Keuls).
The miR-223 locus is located on the X chromosome with wild-type (miR-223 +/y ) and miR-223 knockout (miR-223 −/y ) littermates being available. 23 In miR-223 +/y mice, hypoxia increased the number of fully muscularized pulmonary arteries, an effect more pronounced in miR-223 −/y mice ( Figure 1 F ) as were the hypoxia-induced increase in right-ventricular systolic pressure (RVSP) ( Figure 1 G ) and RV hypertrophy ( Figure 1 H ). Interestingly, hypoxia-induced changes in RV function, with decreased cardiac output ( Figure 1 I ) and impaired contractility; assessed as increased myocardial performance index (MPI; Figure 1 J ), were also more pronounced in the miR-223 −/y littermates.
Granulocytes lacking miR-223 are hyper-mature and the lungs of adult (1.2 years) miR-223 −/y animals display inflammation. 23 This phenomenon was not observed in the present study in which significantly younger animals were studied (11 weeks). However, because of this potential complication, experiments were repeated using wild-type mice treated with antagomirs directed against miR-223. The latter penetrated into the lung and slightly increased arteriolar muscularization under normoxic conditions ( Figure 2 A and B ). In combination with hypoxia, the effects on lung morphology were more pronounced in miR-223 antagomir-treated animals ( Figure 2 C ). The miR-223 inhibitor also penetrated the RV, increased hypoxia-induced RV hypertrophy and functional impairment ( Figure 2 D–I ), and increased the expression of hypertrophy-related genes ( Figure 2 J–L ).
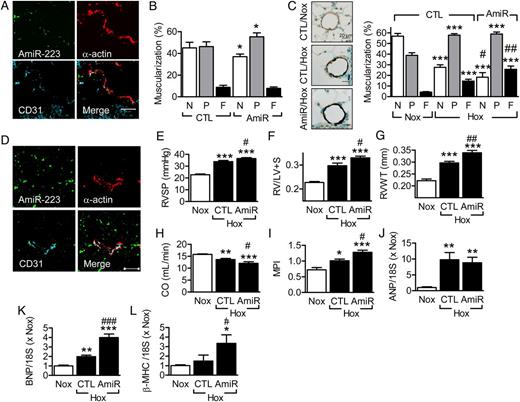
Effect of miR-223 inhibition on pulmonary muscularization, right heart hypertrophy and function. ( A ) Cy3-labelled antagomir-223 (AmiR-223) distribution in mouse lung 2 days after administration (8 mg/kg i.v.); bar = 50 μm. Comparable results were obtained in two additional experiments. ( B ) Muscularization of small diameter (20–70 µm) pulmonary arteries in lungs from mice maintained in normoxic conditions and treated with control (CTL) or antagomir-223 (AmiR). N = non-muscularized; P = partially muscularized; F = fully muscularized ( n = 4–5). ( C ) Effect of hypoxia, CTL antagomir, or antagomir-223 on α-actin staining in lung sections and pulmonary artery (20–70 µm) muscularization ( n = 6–8). ( D–L ) Effect of antagomir-223 on hypoxia-induced changes in the right heart. ( D ) Cy3-labelled antagomir-223 (AmiR) distribution in the right ventricle 2 days after administration; bar = 50 μm. Similar results were obtained in two additional animals. ( E–L ) Effect of a control (CTL) antagomir vs. antagomir-223 (AmiR) on ( E ) right-ventricular systolic pressure (RSVP), ( F ) RV hypertrophy (RV/LV+S), ( G ) right-ventricular wall thickness (RVWT), ( H ) cardiac output (CO), ( I ) myocardial performance index (MPI), ( J ) atrial natriuretic peptide (ANP), ( K ) brain natriuretic peptide (BNP), and ( L ) β-myosin heavy chain (β-MHC). Data from animals maintained under normoxic (Nox) conditions were included for reference. The bar graphs summarize data obtained with 7–8 ( E–I ) or 5 ( J–L ) animals in each group. * P <0.05, ** P <0.01, *** P <0.001 vs. the appropriate CTL/Nox group; #P <0.05, ##P <0.01, ###P <0.001 vs. CTL/Hox, ANOVA (Newman–Keuls).
CCAAT/enhancer binding protein-α (C/EBPα) response elements are contained in the miR-223 core promoter and reported to positively regulate its activity. 32 In line with results showing that C/EBPα is regulated by HIF-1α, 33 hypoxia attenuated C/EBPα expression in murine lungs and in COS-7 cells ( Supplementary material online, Figure S1A and B ). While the overexpression of C/EBPα in COS-7 cells increased miR-223 promoter activity ( Supplementary material online, Figure S1C ), it failed to rescue promoter activity in cells exposed to hypoxia, even though cellular levels of the overexpressed C/EBPα remained constant ( Supplementary material online, Figure S1D ). This could be explained by the binding of HIF-1α to C/EBPα which alters the function of the latter. 34 Indeed, expression of a constitutively active HIF-1α mutant decreased basal activity of the miR-223 promoter and abolished the enhancing effect of C/EBPα ( Supplementary material online, Figure S1E ), a similar effect was observed in cells overexpressing HIF-2α (data not shown).
3.2 miR-223 in the right heart
Given the clear effects of miR-223 deletion and inhibition on RV function, we took a closer look at miR-223 levels in the heart. MiR-223 expression was markedly stronger in the RV than the LV ( Figure 3 A and B ). Moreover, hypoxia significantly decreased miR-223 levels in the RV without affecting its expression in the LV ( Figure 3 B ). Given that hypoxia induced changes in the expression of a range of miRNAs in the lung, we determined the importance of miR-223 in the regulation of RV function using an adeno-associated virus rescue approach. MiR-223 (or EGFP as a control) under the control of the cardiac troponin T type 2 promoter was targeted effectively to cardiac myocytes ( Figure 3 C and D ) and not the lung. Compared with animals treated with EGFP viruses, the miR-223 rescue improved cardiac output and MPI values in animals exposed to hypoxia ( Figure 3 E and F ).
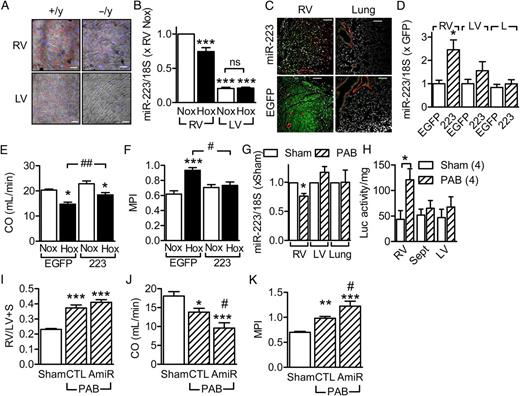
Role of miR-223 in the regulation of right heart function. ( A ) Representative in situ hybridizations showing miR-223 (red) expression in the right and left ventricles from wild-type (+/y) and miR-223 −/y littermates; blue = nuclei, bar = 20 µm. Comparable results were obtained in two additional experiments. ( B ) Comparison of miR-223 expression in the right (RV) and left (LV) ventricles from wild-type mice maintained under normoxic conditions (Nox) or exposed to hypoxia (Hox), n = 6. ( C ) Representative images showing effectiveness of cardiac myocyte transduction with AAV-EGFP, the miR-223 groups are shown as a control; bar = 100 µm. Comparable results were obtained in three additional experiments. ( D ) Efficiency of the miR-223 rescue in the right ventricle (RV), left ventricle (LV), and lung ( n = 5–7). ( E and F ) Effect of adeno-associated virus transduction of EGFP or miR-223 on hypoxia-induced changes in ( E ) cardiac output (CO, mL/min) and ( F ) myocardial performance index (MPI), ( n = 5–8). ( G ) MiR-223 levels in the right ventricle (RV), left ventricle (LV), and lungs of wild-type mice subjected to pulmonary artery banding (PAB), ( n = 5–6). ( H ) Luciferase activity in tissues isolated from HIF1α ODD-luciferase reporter mice 3 weeks after either sham treatment or PAB ( n = 4). ( I–K ) Effect of miR-223 down regulation on PAB-induced right heart failure; ( I ) RV/(LV+S), ( J ) cardiac output (CO, mL/min), and ( K ) myocardial performance index (MPI), ( n = 11–12). * P <0.05, ** P <0.01, *** P <0.001 vs. EGFP, Nox or sham; #P <0.05, ##P <0.01, ###P <0.001 vs. EGFP/Hox or CTL antagomir, ANOVA (Newman–Keuls).
To determine whether the changes in miR-223 expression in the RV could also be elicited by increased mechanical load, mice were subjected to pulmonary artery banding. Banding alone decreased the expression of miR-223 in the RV but had no effect on its expression in either the LV or lung ( Figure 3G ). Studies with HIF-1α ODD-luciferase reporter mice revealed that banding resulted in RV ischaemia/hypoxia ( Figure 3H ), with no detectable change in luciferase activity in LVs or lungs from the same animals. Banding also resulted in RV hypertrophy ( Figure 3I ) and impaired RV function ( Figure 3 J and K ), all of which were significantly poorer in animals additionally treated with antagomirs directed against miR-223. These data indicate that pulmonary artery banding attenuates right-ventricular miR-223 levels by inducing local hypoxia and, as with the chronic hypoxia model, decreased miR-223 levels attenuate right heart function.
3.3 IGF1R is targeted by miR-223
The insulin-like growth factor-I receptor (IGF-IR) is a well characterized target of miR-223 23 , 35 , 36 and mutation of the seeding sequence in the 3′ untranslated region prevents of IGF-IR prevents its regulation ( Figure 4A ). Consistent with its effects on miR-223 levels, hypoxia increased IGF-IR expression in lungs and RV of mice kept in hypoxic conditions ( Figure 4B–D ). Antagomir-223 and miR-223 deletion reproduced the effects of hypoxia on IGF-IR in the lung ( Figure 4 E and F ). In human samples, PH was associated with decreased expression of miR-223 (cycle threshold value, Figure 4 G ) and a concomitant increase in the expression of IGF-IR ( Figure 4 H ). Pulmonary artery banding in mice also increased IGF-IR expression in the RV but not the LV ( Figure 4 I ).
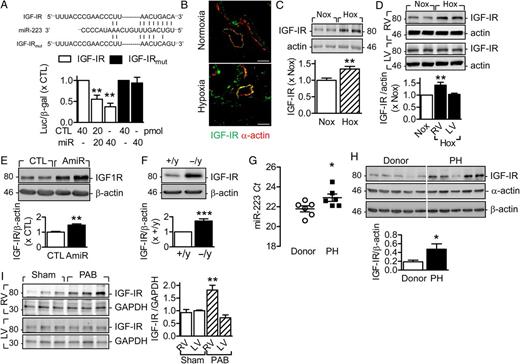
MiR-223 targets IGF-IR and regulates its signalling. ( A ) Effects of a control miRNA and miR-223 on the activity of a wild-type and mutated IGF-IR 3′UTR reporter gene in COS7 cells. The upper sequence shows the predicted miR-223 binding sequence within the IGF1R 3′ UTR and the lower one shows the mutated form used in the functional studies. The middle sequence is that of miR-223 ( n = 3). ( B and C ) IGF-IR expression in lungs from wild-type mice maintained under normoxia (Nox) or exposed to hypoxia (Hox) for 3 weeks as determined by immunohistochemistry; bar = 50 µm. Comparable results were obtained in two additional experiments ( B ), and western blotting ( n = 10) ( C ). ( D ) Effect of hypoxia on IGF-IR expression in the right ventricle (RV) and left ventricle (LV), ( n = 6). ( E ) Effect of antagomir-223 (AmiR) vs. control (CTL) antagomir on pulmonary IGF-IR expression ( n = 4). ( F ) Comparison of IGF-IR levels in lungs from miR-223 +/y and miR-223 −/y littermates ( n = 4). ( G and H ) MiR-223 levels ( G ), and IGF-IR expression ( H ) in lung tissue from human donors and patients with idiopathic pulmonary artery hypertension (PH); ( n = 5–6). ( I ) Effect of pulmonary artery banding (PAB) on IGF-IR expression in the right ventricle (RV) and left ventricle (LV), ( n = 5–9). * P <0.05, ** P <0.01, *** P <0.001 vs. CTL, Nox, +/y, donor or RV/Sham, ANOVA (Newman–Keuls).
Next the consequences of modifying miR-223 levels on IGF-IR expression in murine pulmonary vascular smooth muscle cells were assessed. Although miR-223 was readily detectable in freshly isolated cells, levels declined markedly (over 99%) in culture, a phenomenon previously reported for endothelial cells. 37 Correspondingly, basal IGF-IR expression in cultured smooth muscle cells was high ( Supplementary material online, Figure S2A ). Transfection with pre-miR-223 decreased the expression of the IGF-IR protein, and attenuated IGF-IR signal transduction, as assessed by the IGF-I-induced phosphorylation of Akt ( Supplementary material online, Figure S2B ). Pre-miR-223 overexpression also significantly attenuated basal proliferation and abolished the response to IGF-I ( Supplementary material online , Figure S2C ). Because of low miR-223 levels in the cells used there was no effect of miR-223 antisense oligonucleotides on IGF-IR expression or Akt phosphorylation. Pre-miR-223 transfection also resulted in a doubling of cell apoptosis under serum-free conditions ( Supplementary material online, Figure S2D ) and increased the expression of the pro- (154 ± 44%) and active (158 ± 22%) forms of caspase 3 at the same time as decreasing BCL2 expression (64 ± 15%, Supplementary material online, Figure S2E ). Thus a decrease in miR-223 could account for the enhanced pulmonary muscularization observed in miR-223 −/y mice and mice treated with the miR-223 antagonist.
3.4 Therapeutic benefit of IGF-IR deletion and inhibition
Given that these data implied that the protective effects of miR-223 on RV function may be related to decreased IGF-IR expression, further studies employed experiments animals in which the IGF-IR was inactivated in adult cardiac myocytes by a tamoxifen-inducible Cre recombinase (iCMIGF-IRKO mice). 24 Tamoxifen application resulted in a significant depletion of the IGF-IR from cardiac myocytes ( Figure 5 A ) and the hypoxia-induced up-regulation of IGF-IR in the RV was prevented ( Figure 5 B ). The cardiac myocyte-specific down-regulation of the IGF-IR attenuated the increase in RV mass induced by hypoxia ( Figure 5 C ); and improved the RV functional parameters ( Figure 5 D and E ). Similar benefits of IGF-IR down-regulation were evident in animals subjected to pulmonary artery banding ( Figure 5 F–H ).
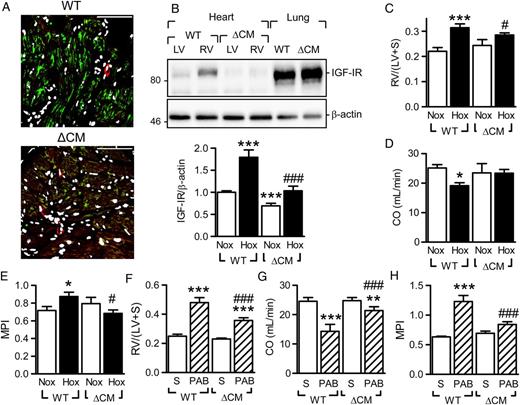
Effect of cardiac myocyte specific IGF-IR deletion on hypoxia-induced changes in right-ventricular structure and function. ( A ) IGF-IR (green) levels in tamoxifen-treated Cre- (WT) and Cre+ (ΔMC) iCMIGF-IRKO littermates; vessels were labelled with α-actin (red) and nuclei with DAPI (white), bar = 50 µm. Comparable results were obtained in two additional experiments. ( B ) IGF-IR levels in right (RV) and left (LV) ventricles from tamoxifen-treated Cre- (WT) and Cre+ (ΔMC) iCMIGF-IRKO littermates subjected to hypoxia for 3 weeks. Samples from lungs were included to demonstrate that the knockout was cardiac specific ( n = 5–8). ( C – E ) Consequences of hypoxia and IGF-IR cardiomyocyte deletion on cardiac structure and function; ( C ) RV/(LV+S), ( D ) cardiac output (CO, mL/min) and ( E ) myocardial performance index (MPI). ( F–H ) Consequences of IGF-IR cardiomyocyte deletion on cardiac structure and function following sham treatment (S) or pulmonary artery banding (PAB); ( F ) RV/(LV+S), ( G ) CO (mL/min), and ( H ) MPI. The bar graphs summarize data obtained with 5–8 ( C–E ) or 6–7 ( F–H ) animals in each group. * P <0.05, ** P <0.01, *** P <0.001 vs. the corresponding Nox or sham group; #P <0.05, ##P <0.01, ###P <0.001 vs. WT/Hox or WT/PAB, ANOVA (Newman–Keuls).
Considering that the AAV-mediated transduction of cardiomyocytes is a rather complex therapeutic option, we determined whether the IGF-IR inhibitor, GSK1904529A, 27 could attenuate the pulmonary remodelling and cardiac dysfunction associated with chronic hypoxia. To imitate therapy conditions more closely, inhibitor treatment was started 1 week after initiation of hypoxia and given daily for the remaining 2 weeks. As before, hypoxia increased the muscularization of small pulmonary arteries in vehicle-treated animals but the increase in fully muscularized vessels was blunted by the inhibitor ( Figure 6 A ). The decrease in pulmonary remodelling was paralleled by an improvement in RVSP ( Figure 6 B ) and RV hypertrophy ( Figure 6 C). In animals subjected to pulmonary artery banding, GSK1904529A had no effect on the RV pressure which was prefixed by the banding procedure ( Figure 6 D ), but decreased RV hypertrophy ( Figure 6 E ) and improved cardiac function ( Figure 6 F and G ). The IGF-IR inhibitor also attenuated the increase in hypertrophy markers induced by pulmonary banding ( Figure 6 H ), at the same time as decreasing right-ventricular fibrosis as assessed by the expression of connective tissue growth factor, fibronectin, collagens A1 and A2.
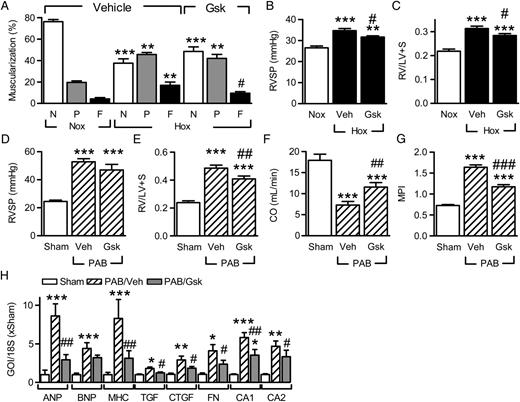
Consequences of IGF-IR inhibition on pulmonary vascular remodelling and right heart function. ( A ) Effect of IGF-IR inhibition on pulmonary vascular muscularization (20–70 μm vessels) in hypoxic (Hox; 10% O 2 , 21 days) mice treated with vehicle (Veh) or GSK1904529A (Gsk; 30 mg/kg p.o.) from Day 7. N = non-muscularized; P = partially muscularized; F = fully muscularized ( n = 5–7). ( B and C ) Effect of IGF-IR inhibition in hypoxia on ( B ) RV systolic pressure (RVSP) and ( C ) right-ventricular hypertrophy (RV/LV+S), ( n = 5–10). ( D–G ) Effect of GSK1904529A on right heart hypertrophy and function following pulmonary artery banding (PAB); ( D ) RVSP, ( E ) RV/LV+S, ( F ) CO, and ( G ) MPI ( n = 5–10). ( H ) Effect of GSK1904529A on the expression of hypertrophy- and fibrosis-related genes of interest (GOI) in animals subjected to PAB ( n = 5). * P <0.05, ** P <0.01, *** P <0.001 vs. Nox or sham; #P <0.05, ##P <0.01, ###P <0.001 vs. Hox/Veh or PAB/Veh, ANOVA (Newman–Keuls).
4 Discussion
The results of this study show that, in an experimental setting of hypoxia- and load-induced PH, miR-223 is co-regulated in the lung vasculature and RV but not regulated in the LV. Moreover, a signalling cascade with hypoxia- and load- or ischaemia-induced loss of miR-223 initiates the enhanced expression of IGF-IR and subsequent IGF-I signalling that contributes to pulmonary smooth muscle cell proliferation, RV hypertrophy, and failure ( Supplementary material online, Figure S3 ).
Several miRNAs are known to be regulated by hypoxia, the best studied is undoubtedly the HIF-1α-regulated miR-210, 30 , 31 , 38 and as expected this miRNA was among the up-regulated miRNAs in the hypoxic mouse lung. An increase in miR-494, a miRNA thought to be critical for myocyte adaptation and survival during hypoxia/ischaemia 39 was also detected, as well as a decrease in the expression of miR-223. MiR-223 was chosen for further study as although it was previously assumed to be restricted to the myeloid compartment of the haematopoietic system, 40 it was clearly expressed in pulmonary vascular smooth muscle cells in vivo and miR-223 −/y mice were more sensitive to hypoxia-induced pulmonary artery remodelling and right-ventricular hypertrophy than their wild-type littermates.
Right heart failure is the cause of death of most patients with severe PH, yet little is known about the cellular and molecular causes of RV failure. Moreover, although several studies have determined changes in the miRNA profile associated with different models of PH in animal models as well as in samples from human patients (for review see Bienertova-Vasku et al . 41 ), few studies have focused on linking changes in the pulmonary system with those taking place in the right heart as a potential link to the development of right heart failure. Extrapolating data from left heart failure is unlikely to provide useful information given the marked differences in the embryonic origin and mechanical stimulation. The latter facts have led to the proposal that a molecular right heart failure programme exists that distinguishes RV failure from adaptive RV hypertrophy, and that also differs from a left heart failure programme. 42 There is circumstantial evidence to support a possible role of cardiac miRNAs is such a programme as several miRNAs, such as miR-143/145, miR-27b, and miR-31, were found to be differentially expressed in the RV and LV. 42 Although miR-223 was not identified in the latter study, clear differences in its expression (RT–PCR and in situ hybridization) exist between the right and left heart with a higher expression in the RV. Also, RV miR-223 expression was attenuated by chronic hypoxia and the function of the right heart in animals exposed to hypoxia was significantly attenuated in animals lacking miR-223. Not only was chronic hypoxia associated with a decrease in miR-223 expression but pressure overload as a consequence of pulmonary artery banding also decreased miR-223 levels. These findings suggest that miR-223 expression is common to the lung and right heart and may represent part of a molecular right heart failure programme.
What mechanism underlies the hypoxia-induced decrease in miR-223 levels? We found that the hypoxia-induced decrease in pulmonary miR-223 levels was linked to the down-regulation of C/EBPα which is similar to the reported regulation of miR-223 in granulocytes 43 , 44 and endothelial cells. 45 However, although the overexpression of C/EBPα increased miR-223 reporter activity under normoxic conditions it failed to prevent the decrease observed following exposure to hypoxia. This observation could not be explained by a down-regulation of the overexpressed protein but seems to be related to the binding of HIF-1α to C/EBPα, an interaction known to alter C/EBPα function. 34 Indeed, the overexpression of a constitutively active HIF-1α mutant was enough to abolish the stimulating effect of C/EBPα on miR-223 promoter activity under normoxic conditions. Although such a mechanism could account for the decrease in pulmonary levels of the miRNA in animals exposed to chronic hypoxia, it seemed unlikely that it could account for the change in miR-223 levels in animal subjected to pulmonary artery banding. However, the latter intervention did induce a detectable hypoxic response in the RV; as assessed using HIF-1α ODD-luciferase reporter mice.
miR-223 was originally predicted to affect approximately 200 proteins in murine neutrophils but the expression of only 78 proteins was detected using SILAC analysis in cells from miR-223 −/− mice. 46 A few of these have been studied in detail; in particular IGF-IR. 23 , 35 , 36 We could confirm the link between miR-223 and IGF-IR expression in the mouse lung as well as in human samples. Indeed, miR-223 expression was significantly decreased while IGF-IR expression was increased in the lung vasculature from patients with primary PH. In addition to the differential expression of miR-223 in the RV vs. the LV observed in this study, a recent report identified spatial asymmetry in IGF-I signalling in RVs and LVs from healthy human hearts. 47
Changes in the expression of one miRNA cannot be expected to mimic the phenotype of the hypoxic lung or the associated changes in right-ventricular function. However, in vivo miR-223 antagonism was sufficient to increase in the expression of IGF-IR and to increase the number of partially muscularized pulmonary arterioles (20–70 µm) as well as to enhance right-ventricular hypertrophy and exacerbate the loss of right-ventricular function in animals exposed to hypoxia as well as in a model of pressure overload. Moreover, in a rescue experiment in which cardiac myocyte expression of miR-223 was increased, the right-ventricular dysfunction associated with chronic hypoxia was attenuated. All of these observations indicate that miR-223 plays a key role in the regulation of the pulmonary-right heart axis.
Each miRNA potentially targets hundreds of target proteins and deciphering which of these can be exploited for potential therapy is a challenge particularly as multiple miRNAs can regulate a single protein. 48 This implies that restoring the levels of only one miRNA may not be expected to fully suppress the elevated IGF-IR expression linked to right heart failure. Therefore, it was necessary to determine how crucial the increase in IGF-IR expression was for the development of PH and right heart dysfunction. This was particularly relevant as IGF-IR is essential for pulmonary and cardiac development 49 and the growth factor is generally classed as a cardio-protective growth factor. 50 , 51 It should, however, be stressed that despite earlier reports, the influence of the IGF-IR on the development of PH and heart failure is unclear as in mice a monoclonal antibody against the IGF-IR improved survival and accelerated resolution of fibrosis after bleomycin-induced lung injury. 52 Thus the inactivation of the IGF-IR in adult animals may be beneficial. This investigation made use of both pharmacological as well as genetic approaches to assess the importance of IGF-IR expression on the development of right heart failure. Both approaches improved right heart function in two different models, i.e. hypoxia-induced PH and right heart dysfunction as well as elevated mechanical loading by pulmonary artery banding both of which impacted on the expression of miR-223 and IGF-IR in the RV but not the LV.
Taken together, our findings suggest that the miR-223 - IGF-IR signalling pathway is regulated in parallel in both the lung vasculature and the RV under conditions of hypoxia induced PH. Interference with this pathway in non-aged hearts, as demonstrated here by the use of a genetic inactivation of the growth factor receptor and a selective small-molecule inhibitor of IGF-IR, highlights the early role of the IGF-IR in the right heart failure programme and may lead to the development of novel therapeutic strategies that target the development of PH and right heart failure.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (Exzellenzcluster 147 ‘Cardio-Pulmonary System’), a ‘shared expertise’ grant from the German Centre for Cardiovascular Research (DZHK) (to I.F., H.A.K., O.J.M.), and by a student stipend (to L.S.) from the Frankfurt International Research Graduate School for Translational Biomedicine.
Acknowledgements
The authors are indebted to Isabel Winter and Christina Vroom for expert technical assistance.
Conflict of interest : none declared.
References
Author notes
Both authors contributed equally.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}