





Abstract
Sulfur dioxide (SO2) is an air pollutant that impedes neonatal development and induces adverse cardiorespiratory health effects, including tachycardia. Here, an animal model was developed that enabled characterization of (i) in vivo alterations in heart rate and (ii) altered activity in brainstem neurons that control heart rate after perinatal SO2 exposure.
Pregnant Sprague–Dawley dams and their pups were exposed to 5 parts per million SO2 for 1 h daily throughout gestation and 6 days postnatal. Electrocardiograms were recorded from pups at 5 days postnatal to examine changes in basal and diving reflex-evoked changes in heart rate following perinatal SO2 exposure. In vitro studies employed whole-cell patch-clamp electrophysiology to examine changes in neurotransmission to cardiac vagal neurons within the nucleus ambiguus upon SO2 exposure using a preparation that maintains fictive inspiratory activity recorded from the hypoglossal rootlet. Perinatal SO2 exposure increased heart rate and blunted the parasympathetic-mediated diving reflex-evoked changes in heart rate. Neither spontaneous nor inspiratory-related inhibitory GABAergic or glycinergic neurotransmission to cardiac vagal neurons was altered by SO2 exposure. However, excitatory glutamatergic neurotransmission was decreased by 51.2% upon SO2 exposure. This diminished excitatory neurotransmission was tetrodotoxin-sensitive, indicating SO2 exposure impaired the activity of preceding glutamatergic neurons that synapse upon cardiac vagal neurons.
Diminished glutamatergic, but unaltered inhibitory neurotransmission to cardiac vagal neurons provides a mechanism for the observed SO2-induced elevated heart rate via an impairment of brainstem cardioinhibitory parasympathetic activity to the heart.
1. Introduction
The 1952 London Smog incident resulted in over 12 000 deaths due to elevated ambient air pollution levels and was one of the principal events to shed light on the deleterious effects of air pollution on human health.1,2 In 2002, the World Health Organization estimated that 3 million people die annually from the combined health effects of exposure to polluted air, despite the existence of clean air regulations around the world.3 Of the six defined air pollutants in the USA [carbon monoxide, lead, nitrogen oxides, ozone (O3), particulate matter (PM), and sulfur dioxide (SO2)], the roles of O3 and PM in disrupting cardiorespiratory function have been well documented.4 Although studies have shown an increased risk for cardiorespiratory-related death following exposure to SO2, the mechanism(s) responsible, as well as the site(s) of action, have not been thoroughly investigated.5,6
SO2 is a gaseous air pollutant that is readily solubilized upon entering the airways, dissociating into its derivatives, bisulfite and sulfite, triggering free radical formation, and inducing tissue damage and bronchoconstriction.7 In addition to pulmonary toxicity, exposure to SO2 has many known cardiovascular effects including hypertension,8 tachycardia or a decrease in the inter-beat interval of heart rate (HR),9,10 myocardial infarction, and cardiac-related morbidity and mortality.4 While it has been suggested that some of these effects may result from inflammatory events following inhalation, another likely possibility is that SO2 alters cardiorespiratory activity by varying cardiac function through alterations to the autonomic nervous system. Indeed, SO2 exposure elevates HR and induces autonomic imbalance to the heart, particularly by diminishing parasympathetic cardioprotective activity.10,11
While known adverse physiological effects of SO2 exposure have been examined primarily in adult populations, the Environmental Protection Agency has stated that neonates, a particularly sensitive subpopulation, have so far been ignored in physiological studies.12 Previous work has linked prenatal air pollution exposure to several adverse birth outcomes including intrauterine growth restriction, very low birth weight, and congenital heart defects.13–15 A strong link between SO2 exposure and sudden infant death syndrome (SIDS) has also been shown.16 A peak in pollutant levels during the winter months increased the number of SIDS cases with a 7-week delay suggesting prenatal SO2 exposure increases the risk of SIDs. Other work has confirmed the finding that SO2 exposure is correlated with SIDS and showed an 8.49% increase in the incidence of SIDS for every inter-quartile increase in SO2 levels.17 Given the impacts of SO2 on the cardiorespiratory system, and that SIDS results from cardiopulmonary collapse, research examining the impact of perinatal SO2 exposure on cardiorespiratory function is highly warranted.
Owing to the lack of focus on neonatal cardiovascular health, we herein developed a novel animal model of perinatal SO2 exposure and characterized the in vivo effects of SO2 on parasympathetic control of HR, as well as alterations in the cellular changes in brainstem neurons that control HR, which are a likely target for these adverse cardiovascular consequences.
2. Methods
Experiments were performed on Sprague–Dawley rats (Hilltop Lab Animals, Inc., Scottdale, PA, USA) housed in The George Washington University (GWU) animal care facility. All animal procedures were approved by the GWU Institutional Animal Care and Use Committee, and all procedures are in accordance with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association and the National Institutes of Health publication Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011). Rats were maintained under standard environmental conditions (12:12 h light:dark cycle) with free access to food and water.
2.1 SO2 exposure protocol
Starting on the first day of gestation through 6 days postnatal (P6), pregnant rats (and their pups) were exposed to 5 parts per million (p.p.m.; 13.1 mg/m3) SO2 for 1 h daily. This concentration was selected based on previously published studies in which 5–10 p.p.m. was selected to mimic urban atmospheres,7,18 as well as the Occupational Safety and Health Administration Short-Term Exposure Limit (STEL) of 5 p.p.m., which is the maximum 1 h personal exposure limit.19 A Single Toxic Gas Monitor for SO2 (RKI Instruments, Union City, CA, USA) was used to monitor the concentration of SO2 inside a sealed chamber (E-Z Systems, Palmer, PA, USA). In control experiments the same exposure protocol was used except animals were exposed to medical-grade air. SO2 and medical-grade air tanks were purchased from Roberts Oxygen (Rockville, MD, USA).
2.2 Unrestrained ECG recording
Three standard electrocardiogram (ECG) electrodes were affixed to unanaesthetized and unrestrained P5 pups in the Lead II configuration, and ECG activity was amplified (CWE, Inc., Ardmore, PA, USA), digitized and recorded using the DSI Ponemah software (St Paul, MN, USA). The effect of SO2 exposure on basal and reflex control of HR was examined by eliciting the diving reflex, a powerful parasympathetic reflex resulting in bradycardia,20 by placing one to two drops of cold (10°C) water on the pup's nose. Atropine sulfate (1 mg/kg, sc)21 was subsequently injected to block parasympathetic activity, and the reflex was retested. Atenolol (10 mg/kg, sc)21 was then administered to additionally block the sympathetic control of HR to determine intrinsic HR. Pups were kept on a heating pad to ensure stable body temperature at 34°C. Atropine sulfate and atenolol were received from Sigma-Aldrich (St Louis, MO, USA). None of the pups used for ECG recordings underwent any surgical procedures.
2.3 Labelling of cardiac vagal neurons and slice preparation
In a separate group of animals, pups at P1 were anaesthetized via hypothermia and cooled to ∼4°C. Once HR significantly slowed and no pain reflex was elicited, a right thoracotomy was performed to expose the heart, and the retrograde tracer rhodamine (XRITC, Invitrogen, 2% solution, 20–50 μL) was injected into the pericardial sac to label cardiac vagal neurons (CVNs). Following the surgery, buprenorphine (0.1–0.5 mg/kg) was immediately administered sc and pups were warmed to raise their body temperatures on a heating pad. Pups were monitored for 30 min and every 20 min thereafter until ambulatory, and two more injections of buprenorphine were given within the next 2–12 h.
After 3–5 days of recovery, animals were overdosed with isoflurane and euthanized by cervical dislocation. Brain tissue was collected and placed in 4°C physiological saline buffer solution with the following composition: NaCl (140 mM), KCl (5 mM), CaCl2 (2 mM), glucose (5 mM), and HEPES (10 mM). The medulla was dissected to preserve the hypoglossal cranial rootlets, mounted on a cutting block, and placed into a vibrating blade microtome. A single slice (800 µm) that included the rostral ventral lateral medulla, the hypoglossal nerve rootlet, and the nucleus ambiguus was obtained and submerged in a recording chamber that allowed perfusion above and below the slice with room temperature artificial cerebrospinal fluid with the following composition: NaCl (12 5 mM), KCl (3 mM), CaCl2 (2 mM), NaHCO3 (26 mM), glucose (5 mM), HEPES (5 mM) in equilibrium with 95% O2–5% CO2. The osmolarity of all solutions was 285–290 mOsm, and the pH was maintained between 7.35 and 7.4.
2.4 Recording respiratory network activity and patch-clamp techniques
The thick medullary slice preparation generates rhythmic inspiratory-related motor discharge in hypoglossal cranial nerves.22 Spontaneous respiratory-related activity was recorded by monitoring motorneuron population activity from the hypoglossal rootlets using a suction electrode. Hypoglossal nerve rootlet activity was amplified (50 000×), filtered (10–300 Hz pass; CWE, Inc., Ardmore, PA, USA), and electronically integrated ([tau] = 50 ms; CW).
CVNs were identified by the presence of the fluorescent tracer and differential interference contrast optics along with infrared illumination and infrared-sensitive video detection cameras to gain enhanced spatial resolution. Patch pipettes were filled with a solution at pH 7.3 consisting of either KCl (150 mM), MgCl2 (4 mM), EGTA (10 mM), Na-ATP (2 mM), HEPES (10 mM) or K-gluconic acid (150 mM), HEPES (10 mM), EGTA (10 mM), MgCl2 (1 mM), CaCl2 (1 mM) to isolate for inhibitory or excitatory currents, respectively. Identified CVNs were voltage-clamped at a holding potential of −80 mV.
2.5 Drug application
All drugs used in these experiments to isolate neurotransmission were applied using a pneumatic picopump pressure delivery system (WPI, Sarasota, FL, USA). Drugs used include gabazine (25 µM) to block GABAergic inhibitory neurotransmission, strychnine (1 µM) to block glycinergic inhibitory neurotransmission, and d(−)-2-amino-5-phosphopentanoic acid (AP5; 50 µM) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 50 μM) to block all excitatory neurotransmission.
In experiments where miniature excitatory postsynaptic currents (mEPSCs) were isolated, the voltage-gated sodium channel blocker tetrodotoxin (TTX; 1 μM) was included in the perfusate. All drugs were received from Sigma-Aldrich (St Louis, MO, USA).
2.6 Data analysis
To determine baseline HR, the 15 s prior to diving reflex stimulation was divided into 5 s bins and the average R–R interval for each bin was used to determine HR using the Ponemah Standard ECG Analysis Software (DSI, Minneapolis, MN, USA). The change in HR upon evoking the diving reflex was obtained within 5–10 s after placing one to two drops of cold water on the nose before and after atropine sulfate administration to block parasympathetic activity to the heart. To determine intrinsic HR atenolol was also administered to block sympathetic activity to the heart. Averages for baseline HR, change in HR with and without atropine sulfate, and intrinsic HR were compared between control and SO2-exposed animals. Recordings were considered outliers based upon the very conservative estimation of being >8 SD away from the mean.
Previous work in the laboratory has demonstrated that CVNs exhibit respiratory-related activity via coupling of inhibitory postsynaptic currents (IPSCs) to fictive inspiratory activity recorded from the hypoglossal motor nerve rootlet.23 Before and after each fictive inspiratory burst, 5 s of data were taken and binned into 1 s averages. Data were obtained and then averaged from the first five bursts from each cell.
Spontaneous EPSCs and mEPSCs were recorded for at least 5 min. To determine the frequency of EPSCs, the number of excitatory events was divided by the duration of the recording. Data were obtained and averaged under each condition.
MiniAnalysis (Synaptosoft version 4.3.1) was used to analyse all experimental traces. The threshold for GABAergic, glycinergic, and glutamatergic events was set to five times the root mean square of noise. When multiple cells per animal were used for electrophysiology experiments, the frequency of postsynaptic currents was averaged into a single value for that animal. The averaged data and comparisons between groups included only one data value per animal, with the n-value equal to the total number of animals used for that series of experiments or condition. All data are represented by mean ± SEM. A repeated-measures two-way ANOVA was used to determine statistical significance for the inspiratory-related activity. A Bonferroni post hoc test was used to determine statistically significant differences with a P-value <0.05. A two-tailed unpaired Student's t-test was used to determine statistical significance for the frequency of excitatory events, as well as HR. An F-test was used to determine whether equal variance or unequal variance was used for the t-test, and statistical significance was determined with a P-value <0.05.
3. Results
3.1 Perinatal SO2 exposure elevates HR and blunts reflex activation of parasympathetic activity
To determine the effect of perinatal SO2 exposure on baseline HR, ECGs were recorded from P5 rat pups, as shown in Figure 1. HR, recorded from unanaesthetized and unrestrained pups, was significantly elevated (P < 0.05) following SO2 exposure (424 ± 9 b.p.m.; n = 10) compared with control animals (374 ± 12 b.p.m.; n = 11), an increase of 13.4% (Figure 1B). To determine whether or not reflex changes in parasympathetic activity to the heart were altered by SO2, ECGs were recorded during the activation of the diving reflex, which is a powerful parasympathetic reflex that results in bradycardia after cold water has been placed on the pup's nose (Figure 1C).20 Here, the activation of the diving reflex produced a robust decrease in HR of 43 ± 6 b.p.m. (P < 0.05; n = 11). Atropine sulfate, a muscarinic receptor antagonist, blocked the diving reflex as exhibited by a decrease in HR of only 2 ± 1 b.p.m. (P > 0.05; n = 11). However, in SO2-exposed animals, diving reflex activation decreased HR by only 18 ± 6 b.p.m. (P > 0.05; n = 10), a significant decrease in reflex-evoked changes in the HR, which was also abolished following atropine sulfate injection (2 ± 1 b.p.m.; P > 0.05; n = 10). Together, these data indicate that perinatal SO2 exposure produces a blunting of reflex control of parasympathetic activity (Figure 1E). Additionally, isolation of intrinsic HR, following combined injection of atropine sulfate and the β1-antagonist atenolol, showed that perinatal SO2 also significantly elevated intrinsic HR (266 ± 7 b.p.m.; n = 10) compared with control animals (208 ± 10 b.p.m.; n = 11; P < 0.05; Figure 1D), suggesting an additional mechanism by which SO2 results in an elevated HR.
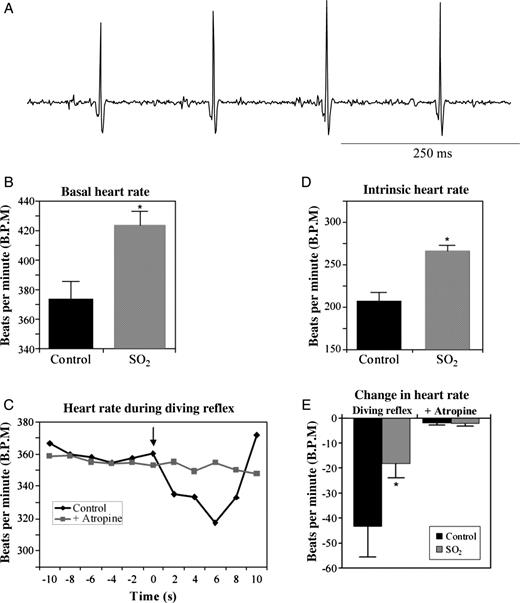
Representative ECG trace recorded from a P5 rat pup (panel A). R–R waves from ECGs were used to determine baseline heart rate (B). SO2-exposed animals exhibited a 13.4% increase in the basal heart rate compared with control pups (SO2, 424 ± 9 b.p.m.; n = 10; control, 374 ± 12 b.p.m.; n = 11; *P < 0.05). The diving reflex was used to examine the reflex control of parasympathetic activity to the heart in P5 pups (C). The basal HR was significantly decreased upon the activation of the diving reflex by placing one to two drops of cold (10°C) water on the pup's nose (indicated by the arrow; black line). Following blockade of parasympathetic activity by administering the muscarinic antagonist atropine sulfate (1 mg/kg, sc), cold water was no longer able to elicit a decrease in HR (grey line). Individual R–R intervals recorded from an example ECG were calculated, converted to HR, and averaged for every 2 s. This response was compared between control and SO2-exposed pups (E). While the diving reflex significantly decreased HR in control animals (−43 ± 6 b.p.m.; n = 11; P < 0.05), the response was significantly blunted in SO2-exposed pups (−18 ± 6 b.p.m.; n = 10; P > 0.05), and abolished by atropine sulfate. The intrinsic HR was isolated by administering atenolol (10 mg/kg, sc) after atropine sulfate (D). SO2-exposed animals exhibited a significantly increased intrinsic HR (266 ± 7 b.p.m.; n = 10) compared with control pups (208 ± 10 b.p.m.; n = 11; *P < 0.05).
Litters were also observed for changes in litter size and average weight at birth. The average litter size for control litters was 13.8 ± 1.3 pups, and for SO2-exposed litters were 14.1 ± 1.0 pups; these differences are not significant. The average weight at birth for control pups was 5.9 ± 0.2 g, and for SO2-exposed pups was 6.2 ± 0.3 g. These differences were also not significant.
3.2 Perinatal exposure to SO2 does not alter inhibitory neurotransmission to CVNs
The cardiovascular effects of SO2 exposure in humans have been hypothesized to be due to a loss of vagal, or parasympathetic, activity.10 The in vivo recordings from our rat model indicate SO2 exposure mimics the cardiovascular changes seen in epidemiology studies, suggesting that this is a viable model to examine the mechanisms that underlie the diminished parasympathetic drive to the heart that occurs with SO2 exposure. Parasympathetic activity to the heart originates from premotor parasympathetic neurons in the brainstem; since these neurons are inherently silent, the synaptic activity to these neurons is a critical determinant of their activity. To determine the mechanism(s) responsible for altered excitability, neurotransmission to premotor CVNs that control HR was studied. We tested whether the diminished parasympathetic activity to the heart was due to an increase in inhibitory and/or decrease in excitatory neurotransmission to CVNs. The following electrophysiological experiments were obtained from a total number of 61 pups from 15 dams (controls: 28 pups from 8 dams; SO2 exposures: 33 pups from 7 dams).
Previous work has demonstrated that CVNs possess respiratory-related patterning consisting of increases in both GABAergic and glycinergic IPSCs during inspiratory activity recorded from the hypoglossal (XII) rootlet.23 Upon isolation of GABAergic IPSCs (Figure 2), perinatal exposure to SO2 was found to have no significant (P > 0.05) changes in GABAergic neurotransmission to CVNs during either spontaneous (control, 6.4 ± 0.9 Hz, n = 6 animals; SO2, 6.9 ± 0.8 Hz, n = 5 animals) or inspiratory-related (control, 11.1 ± 1.3 Hz; SO2, 11.9 ± 0.7 Hz) neurotransmission. Similarly, glycinergic IPSCs also showed no significant (P > 0.05) differences in IPSC frequency between control and SO2-exposed pups during both spontaneous (control, 7.2 ± 1.0 Hz, n = 4 animals; SO2, 6.1 ± 0.8 Hz, n = 5 animals) and inspiratory-related (control, 11.9 ± 1.3 Hz; SO2, 11.6 ± 1.5 Hz) activity, as shown in Figure 3.
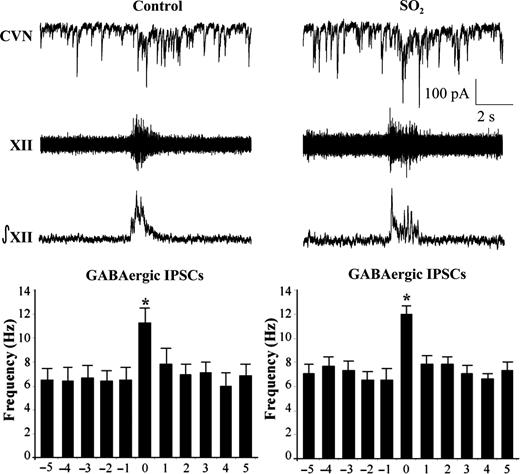
The bursting activity of fictive inspiration was recorded from the hypoglossal rootlet (XII), electronically integrated (∫XII), and recorded simultaneously with GABAergc IPSCs in CVNs (top traces). While there was a significant increase in IPSC frequency during the inspiratory period, there were no differences between control and SO2-exposed pups in either the spontaneous (control, 6.4 ± 0.9 Hz; SO2, 6.9 ± 0.8 Hz) nor inspiratory phases (control, 11.1 ± 1.3 Hz; SO2, 11.9 ± 0.7 Hz), as shown in the histogram (control, n = 6 animals; SO2, n = 5 animals; *P < 0.05).
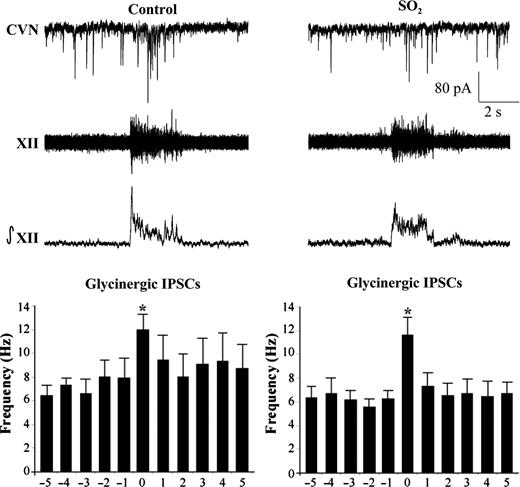
The bursting pattern of the hypoglossal rootlet (XII) was electronically integrated (∫XII) and glycinergic IPSCs were recorded simultaneously in CVNs (top traces). There was a significant increase in IPSC frequency during the inspiratory phase in both conditions; however, there were no differences between control and SO2-exposed animals during either the spontaneous (control, 7.2 ± 1.0 Hz; SO2, 6.1 ± 0.8 Hz) nor inspiratory periods (control, 11.9 ± 1.3 Hz; SO2, 11.6 ± 1.5 Hz), as shown in the histogram (control, n = 4 animals; SO2, n = 5 animals; *P < 0.05).
3.3 Perinatal SO2 exposure alters excitatory signalling to CVNs
While inhibitory neurotransmission to CVNs is known to mediate respiratory sinus arrhythmia, excitatory neurotransmission does not exhibit inspiratory-related coupling. Instead, glutamatergic activity to CVNs is thought to be responsible for generating the basal activity in parasympathetic premotor CVNs and HR changes that occur upon the activation of chemoreceptor and baroreceptor reflexes.24 Spontaneous glutamatergic EPSCs were significantly (P < 0.05) diminished in SO2-exposed animals (2.1 ± 0.3 Hz, n = 11 animals) compared with control (4.3 ± 0.8 Hz, n = 5 animals) pups, a decrease of 51.2%, as shown in Figure 4A and B.
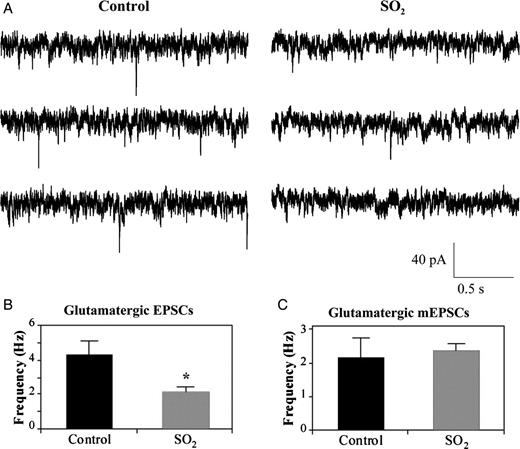
Spontaneous glutamatergic EPSCs (A) were isolated and recorded from labelled CVNs in the nucleus ambiguus using the whole-cell patch-clamp configuration. Representative traces shown are from three consecutive time periods recorded from one control and one SO2-exposed pup, as labelled. EPSC frequency was significantly decreased by 51.2% following perinatal SO2 exposure (2.1 ± 0.3 Hz, n = 11 animals) compared with control pups (4.3 ± 0.8 Hz, n = 5 animals), as shown in (B). Bath application of tetrodotoxin (TTX), a voltage-gated Na+-channel antagonist, was used to record glutamatergic miniature EPSCs (mEPSCs). mEPSC frequency in control (2.1 ± 0.6 Hz, n = 5 animals) and SO2-exposed animals (2.3 ± 0.2 Hz, n = 6 animals) were not significantly different (*P < 0.05).
To determine whether this impaired excitatory neurotransmission was due to dysfunction of the presynaptic release of neurotransmitters, or was action potential-dependent, the voltage-gated Na+ channel blocker TTX was included in the perfusate. The resulting mEPSCs in CVNs were recorded, and the prior difference in the frequency of excitatory neurotransmission between the two groups of animals was abolished, as shown in Figure 4C. Instead, mEPSC frequency in control (2.1 ± 0.6 Hz, n = 5 animals) and SO2-exposed animals (2.3 ± 0.2 Hz, n = 6 animals) were not significantly different. These results indicate that the decrease in glutamatergic neurotransmission to CVNs following SO2 exposure is action potential-dependent and is likely due to alterations in the activity of the preceding neurons in the cardiorespiratory network, rather than to changes in the function of the presynaptic glutamatergic terminals surrounding CVNs.
4. Discussion
Neonates are a poorly studied subpopulation in air pollution physiology, despite traditionally possessing increased sensitivity to several toxins. To the best of our knowledge, we have herein developed the first animal model for investigating SO2 toxicity in this age group. As such, there are three main findings in this study: (i) Our novel animal model of SO2 exposure mimics the cardiovascular effects seen in humans and is the first to focus solely on the effects of perinatal exposure. (ii) While inhibitory neurotransmission to CVNs was unchanged, SO2 significantly reduced glutamatergic EPSC frequency in a TTX-dependent manner, indicating the site of action of SO2 exposure is likely on excitatory glutamatergic neurons and pathways prior to CVNs in the brainstem cardiorespiratory network. (iii) Air pollution exposure is often associated with respiratory disease, but results presented here suggest additional targets, particularly autonomic parasympathetic activity within the brainstem.
This study has shown that perinatal exposure to SO2 increases baseline HR, which in combination with the blunted reflex control of parasympathetic activity demonstrated in the in vivo experiments, elevates the risk of cardiovascular death among a traditionally at-risk subpopulation. Given that SO2 is associated with cardiorespiratory morbidity and mortality in adult populations,4 the physiological data shown here may provide a mechanistic link between SO2 exposure and adverse cardiovascular events. While further work is necessary to identify the alterations in the reflex pathways to CVNs, the results in this study lay the foundation for understanding the diminished parasympathetic activity to the heart in neonates following SO2 exposure.
Several epidemiological studies have examined the effects of SO2 on the adult population, leading to a number of hypotheses about how SO2 alters parasympathetic regulation of the heart. However, the lack of an animal model has hindered further investigation into the mechanism(s) disrupting cardiovascular regulation following exposure. The goal of this study was to test and develop an animal model that would not only facilitate research into the cellular changes arising from SO2 exposure, but would also address the question of whether or not physiological activity is altered by perinatal exposure. In this animal model, Sprague–Dawley rats were exposed during gestation and through 6 days postnatal to 5 p.p.m. SO2 for 1 h daily. The results of this study show SO2 exposure elicits an increase in basal HR, a loss of parasympathetic drive to the heart, and an increase in intrinsic HR. Increases in HR and decreases in cardioprotective parasympathetic activity increase susceptibility to arrhythmia development and cardiovascular disease.25–30 These alterations are likely responsible for many cardiovascular-related deaths and adverse events following SO2 exposure worldwide.
Using the perinatal model developed in this study, we applied in vitro electrophysiology techniques to elucidate a novel mechanism of action for SO2 exposure in brainstem cardiovascular function. Parasympathetic regulation of HR is mediated by CVNs originating in the nucleus ambiguus, and is modulated by both inhibitory and excitatory neurotransmission to these cells.31 Previous work has shown that IPSCs are responsible for inspiratory-related changes in parasympathetic activity to the heart,23 while EPSCs are essential for generating on-going tonic parasympathetic activity.24 Based on our in vivo results, and similar findings by others, we hypothesized SO2 would diminish inspiratory-related inhibitory inputs to CVNs but were surprised to find no differences in either the spontaneous or inspiratory-related activity of neither GABAergic nor glycinergic IPSCs between control and SO2-exposed animals. However, SO2 exposure did significantly diminish glutamatergic EPSCs in CVNs. This loss of excitatory neurotransmission to CVNs would be responsible for a withdrawal of parasympathetic cardioinhibitory vagal activity to the heart, resulting in an elevated basal HR, both of which were present in in vivo recordings from P5 pups.
TTX blocks the activity of voltage-gated Na+ channels, preventing the propagation of action potentials along the axon and isolating action potential-independent mEPSCs, which are thought to be spontaneous release of neurotransmitter from vesicles at presynaptic terminals. The results presented here show mEPSCs in CVNs were not different in animals exposed to SO2, indicating SO2 exposure likely decreases activity of preceding glutamatergic neurons that project to CVNs rather than modifying the presynaptic function. Because SO2 is an inhaled pollutant known to induce bronchoconstriction in animal models of chronic bronchitis,32,33 we suggest that future studies should consider testing the hypothesis that SO2 acts on laryngeal receptors via a multisynaptic pathway to reduce glutamatergic activity in CVNs.
In the late 1990s, several epidemiological studies examined the impact of air pollution exposure on various indicators of cardiovascular health; however, most of this research focused predominately on PM and O3. In 1999, Pope et al.34 published a critical finding linking PM exposure to an altered autonomic state, including tachycardia. This work was quickly followed by more research into air pollution epidemiology, and in the same year Peters et al. published results from an episode in Augsburg, Germany, linking elevated ambient pollution levels to tachycardia in the adult population. These initial studies have been followed by several others examining air pollution events and their impact on measures of cardiovascular health.9,35 It is clear that at least one mechanism by which SO2 exposure disrupts cardiovascular function is through autonomic dysregulation; however, it is still unclear whether these effects are due to an inflammatory response or to stimulation of airway receptors.
A 2006 study by Routledge et al.10 aimed to answer this question using short-term exposure to SO2 in healthy humans. A loss of vagal activity and a lack of circulating inflammatory markers 4 h after exposure suggested that SO2 acts on airway receptors to disrupt neurological inputs to the parasympathetic nervous system. This hypothesis has been echoed and further supported by others arguing that work examining the mechanism behind this evolving hypothesis is needed to advance the field.5 The development of the novel animal model here plays a significant role in achieving that objective. Not only has a new tool been developed, but it has also allowed us to identify pivotal foundations underlying the loss of cardioprotective parasympathetic activity following SO2 exposure. Importantly, work shown here also suggests an additional mechanism of SO2 may be responsible for increasing intrinsic HR, which is an area that should be investigated in future studies. In this study, we have shown that upon inhalation SO2 decreases excitatory inputs to CVNs, likely by depressing a multisynaptic pathway, and hypothesize that this is likely done through the activation of reflexes originating from laryngeal airway receptors. The subsequent vagal withdrawal increases HR, placing the individual at risk for sudden cardiac death.35,36
Funding
This work was supported by the National Institutes of Health (HL49965, HL59895, and HL72006 to D.M.); and the GW Institute for Sustainability Research, Education, and Policy Research Award to A.L.W.
Acknowledgements
A.L.W. is a predoctoral student in the Molecular Medicine Program of The Institute for Biomedical Sciences at The George Washington University. This work is from a dissertation to be presented to the above program in partial fulfilment for the PhD degree.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}