





Abstract
Hyperglycaemia (HG) decreases intracellular tetrahydrobiopterin (BH4) concentrations, and this action may contribute to injury during myocardial ischaemia and reperfusion. We investigated whether increased BH4 by cardiomyocyte-specific overexpression of the GTP cyclohydrolase (GTPCH) 1 gene rescues myocardial and mitochondrial protection by ischaemic preconditioning (IPC) during HG through a nitric oxide (NO)-dependent pathway.
Mice underwent 30 min of myocardial ischaemia followed by 2 h of reperfusion with or without IPC elicited with four cycles of 5 min ischaemia/5 min of reperfusion in the presence or absence of HG produced by d-glucose. In C57BL/6 wild-type mice, IPC increased myocardial BH4 and NO concentrations and decreased myocardial infarct size (30 ± 3% of risk area) compared with control (56 ± 5%) experiments. This protective effect was inhibited by HG (48 ± 3%) but not hyperosmolarity. GTPCH-1 overexpression increased myocardial BH4 and NO concentrations and restored cardioprotection by IPC during HG (32 ± 4%). In contrast, a non-selective NO synthase inhibitor NG-nitro-l-arginine methyl ester attenuated the favourable effects of GTPCH-1 overexpression (52 ± 3%) during HG. Mitochondria isolated from myocardium subjected to IPC required significantly higher in vitro Ca2+ concentrations (184 ± 14 µmol mg−1 protein) to open the mitochondrial permeability transition pore when compared with mitochondria isolated from control experiments (142 ± 10 µmol mg−1 protein). This beneficial effect of IPC was reversed by HG and rescued by GTPCH-1 overexpression.
Increased BH4 by cardiomyocyte-specific overexpression of GTPCH-1 preserves the ability of IPC to elicit myocardial and mitochondrial protection that is impaired by HG, and this action appears to be dependent on NO.
1. Introduction
Hyperglycaemia (HG) commonly occurs in patients with acute myocardial infarction and is associated with poor prognosis and increased mortality.1 The mechanisms responsible for increased morbidity and mortality during HG are unclear; however, disruption of endogenous cardioprotective pathways is likely. For example, ischaemic preconditioning (IPC) is a phenomenon in which brief periods of myocardial ischaemia and reperfusion protect the heart against infarction during a subsequent more prolonged period of coronary artery occlusion.2 We and others have shown that transient HG abolishes the cardioprotective effects of IPC in dogs, rabbits, and in humans,3–6 although the specific mechanisms involved have not been completely elucidated. The present study was designed to address the hypothesis that tetrahydrobiopterin (BH4) is a key mediator of IPC that is adversely modulated by HG, but cardioprotection can be restored by a genetic approach to enhancing BH4 synthesis with cardiomyocyte-targeted overexpression of GTP cyclohydrolase (GTPCH) 1.
BH4 is a reduced unconjugated pterin and essential cofactor that regulates nitric oxide (NO) synthesis by NO synthases (NOS) expressed within cardiomyocytes and coronary vasculature and is an important determinant of NO-dependent signalling.7,8 Increased expression of GTPCH-1, the first and rate-limiting enzyme for the de novo synthesis of BH4, might be a strategy to improve NOS function during HG and IPC.9,10 Endothelium-targeted overexpression of GTPCH-1 by gene transfer or pharmacological supplementation of BH4 maintains endothelial BH4 content and restores the ability of endothelial cells to produce NO.9,11 It remains unknown, however, whether the cardioprotective potential of IPC can be preserved during HG by increasing myocardial BH4 concentrations.
2. Materials and methods
The experimental procedures were approved by the Animal Care and Use Committee of the Medical College of Wisconsin (Milwaukee, WI, USA) and conformed to the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Detailed methods are described in the Supplementary material online.
2.1 Myocardial ischaemia/reperfusion injury in vivo
Myocardial infarction was produced by occluding the left anterior descending coronary artery, as previously described.12–14
2.1.1 Experimental protocol
The effects of HG, IPC, and hyperosmolarity (HOsm) on myocardial ischaemia/reperfusion (I/R) injury were examined in C57BL/6 mice assigned to five experimental groups (7–10 mice/group): control (CON), HG, IPC, IPC + HG, and IPC + HOsm (Figure 1A). All mice were stabilized for 30 min and subjected to 30 min of coronary occlusion followed by 2 h of reperfusion (I/R). IPC was elicited with four consecutive 5 min episodes of coronary occlusion, each followed by a 5 min of reperfusion. d-Glucose (2 g/kg) or mannitol (1.82 g/kg) was administered intraperitoneal 10 min before IPC to produce HG or HOsm, respectively. Tail blood was sampled at baseline, 10 min after d-glucose injection, 15 and 30 min after coronary occlusion, and 60 and 120 min after reperfusion for measurement of blood glucose concentrations (Glucometer). In separate mice, hearts were excised 5 min after reperfusion and myocardium from the area at risk (AAR) was homogenized and centrifuged at 14 000 g for 20 min at 4°C. Tissue NO and its metabolite products (nitrate and nitrite) in the supernatant, collectively known as NOx, were assayed using a NO chemiluminescence analyzer (Siever 280i NO Analyzer).15

Schematic diagram depicting the experimental protocols. (A) Effects of HG, IPC, and hyperosmolarity (HOsm) on infarct size in WT mice subjected to coronary occlusion followed by reperfusion; (B) effects of overexpression of the GTP cyclohydrolase (GTPCH) 1 gene on I/R injury with or without IPC in the presence or absence of HG or l-NAME; (C) effects of GTPCH-1 overexpression on IPC-induced changes in the mitochondrial permeability transition pore (mPTP) in the presence or absence of HG and/or l-NAME.
2.2 Generation of transgenic GTPCH-1 mice
The transgene for the α myosin heavy chain (αMHC) promoter/haemagglutinin (HA) epitope-tag/human GTPCH-1 transgene was kindly provided by Dr Nicholas Alp (University of Oxford, Oxford, UK) for generating cardiomyocyte-targeted overexpression of the human GTPCH-1 gene.16 The transgene was comprised of the murine αMHC promoter including the first three non-translated exons; HA epitope tag; and human GTPCH-1 cDNA; human growth hormone poly A signal. The construct was generated in E. coli cultures by sub-cloning PCR-modified HA-hGTPCH cDNA from phGTPCHA+, into a Sal I restriction site in a plasmid containing the murine αMHC promoter (GenBank Accession Number U71441) within non-translated αMHC exon 3. The resulting plasmid, pαMHCGTPCHA+, was confirmed by DNA sequencing using standard techniques. The human GTPCH-1 transgene was excised from pαMHCGTPCHA+ by NotI digestion, gel-purified, and dissolved in sterile injection buffer (5 mM Tris/Cl, pH 7.5, 0.1 mM EDTA). Oocyte microinjection of the construct was performed at the Transgenic Core Facility of the Medical College of Wisconsin and injected oocytes were transferred into pseudopregnant recipient females. Offspring were screened for genomic DNA from tail tips, using primers specific for the murine αMHC promoter sequence (P1, forward; 5′-CCTTCCTCACCCCCTGGCTTGTCC-3′) and for the HA sequence (P2, reverse; 5′-AGTCGGGCACGTCGTAGGGGTAGG-3′), producing a 516 bp PCR product.
2.3 Expression of GTPCH-1 mRNA in transgenic mice
Total cellular RNA was isolated from heart tissue and complimentary DNA was synthesized from 1 μg of total RNA, as previously described.17 cDNA was used in RT–PCR with the following primers for various GTPCH mRNA transcripts: human (transgenic): forward 5′-CGCCTACTCGTCCATCCTGA-3′, reverse 5′-CCTTCACAATCACCATCTCA-3′ (product size 181 bp); mouse (endogenous): forward 5′-TGCTTACTCGTCCATTCTGC-3′, reverse 5′-CCTTCACAATCACCATCTCG-3′ (product size 181 bp). GAPDH was used as an internal control for all experiments. PCR reactions were performed in a 25 µL volume using GoTaq Green Master Mix (Promega, Madison, WI, USA). The PCR product was resolved by 1% TAE-agarose gel electrophoresis. Densitometric analysis was performed using Alpha Imager (Alpha Innotech Corp., San Leandro, CA, USA).
The effects of overexpressing GTPCH-1 gene on infarct size and IPC-induced cardioprotection were determined in an in vivo model of myocardial I/R injury in the presence or absence of HG (Figure 1B). Transgenic (GTPCH-Tg) mice with cardiomyocyte-specific overexpression of GTPCH 1 and their wild-type (WT) littermate mice were randomly assigned to one of the six groups (n = 7–8 mice/group): CON, NG-nitro-l-arginine methyl ester (l-NAME), IPC, IPC + l-NAME, IPC + HG, IPC + HG + l-NAME. IPC and HG were produced, as described above. l-NAME, a non-selective inhibitor of NOS (1 mg/kg), was administered intraperitoneally 80 min prior to coronary artery occlusion or 40 min prior to IPC. In separate experiments, the effects of GTPCH-1 overexpression on cardiac BH4 and NOx concentrations during IPC were examined in mice randomly assigned to the following four groups: sham, HG, IPC, and IPC + HG. Mice in sham and HG groups underwent no coronary occlusion. Cardiac BH4 concentrations were assayed by high performance liquid chromatography, as described below.
2.4 BH4 assay
BH4 was quantified in left ventricular (LV) biopsies by high performance liquid chromotography with electrochemical detection, as previously described.18 Filtrates were analysed on a high performance liquid chromotography system (ESA Biosciences CoulArray® system, Model 582 and 542) using an analytical Polar-RP column eluted with argon saturated 50 mM phosphate buffer (pH 2.6). Authentic BH4 solutions (10–100 nM) were used as standards and sample concentrations were normalized to protein content measured by the bicinchoninic acid protein assay.
2.5 Transthoracic echocardiography
LV structure and function were evaluated using transthoracic echocardiography.14
2.6 Detection of the mitochondrial permeability transition pore opening in isolated mitochondria
GTPCH-Tg mice and WT littermates were assigned to one of the seven groups: sham, CON, l-NAME, IPC, IPC + l-NAME, IPC + HG, and IPC + HG + l-NAME (Figure 1C). Sham mice were not subjected to coronary occlusion. The hearts were excised after 5 min of final reperfusion, and mitochondria were isolated from the AAR. Opening of the mitochondrial permeability transition pore (mPTP) after in vitro Ca2+ overload was assessed by following changes in the membrane potential (ΔΨm) using the fluorescent dye rhodamine 123 (50 nM; Invitrogen, Carlsband, CA, USA) in the presence of pyruvate and malate (5 mM).14
2.7 Statistical analysis
All data are expressed as mean ± SEM. Statistical analysis was performed with one-way ANOVA followed by the Bonferroni post-hoc test for multiple comparisons of multiple group means or with Student's t-test for comparisons between two group means. Repeated-measures ANOVA was used to compare the differences in heart rate and blood glucose within the groups at different time points. A value of P < 0.05 was considered as statistically significant.
3. Results
3.1 HGinhibited IPC-elicited decreases in infarct size and increases in NO production in WT mice in vivo
Baseline blood glucose was 206 ± 13 mg/dL (n = 31 mice) in unfasted C57BL/6 mice. IPC, mannitol, and I/R had no effect on blood glucose concentrations. Intraperitoneal injection of d-glucose (2 g/kg) resulted in similar increases in blood glucose (Figure 2A) before and after myocardial I/R in WT mice with or without IPC. There were no significant differences in blood glucose concentrations among groups receiving d-glucose (n = 6–9 mice/group, P > 0.05). There were no significant differences in heart rate between experimental groups (Table 1). AAR was not different among the five experimental groups (Figure 2B). IPC significantly decreased myocardial infarct size (Figure 2C) compared with control experiments. HG alone did not significantly alter myocardial infarct size but blocked infarct size reduction produced by IPC.
Heart rate (bpm) during in vivo study
| Group | Baseline | Coronary occlusion | Reperfusion | |||
|---|---|---|---|---|---|---|
| 2 min | 30 min | 60 min | 120 min | |||
| C57BL/6 mice | ||||||
| CON | 404 ± 12 | 411 ± 16 | 425 ± 18 | 413 ± 18 | 401 ± 21 | 391 ± 16 |
| HG | 393 ± 16 | 403 ± 14 | 399 ± 13 | 417 ± 15 | 404 ± 25 | 423 ± 13 |
| IPC | 418 ± 16 | 403 ± 14 | 414 ± 13 | 435 ± 12 | 394 ± 20 | 382 ± 15 |
| IPC+HG | 389 ± 26 | 412 ± 16 | 420 ± 12 | 408 ± 11 | 399 ± 16 | 408 ± 18 |
| IPC + HOsm | 422 ± 18 | 397 ± 19 | 401 ± 21 | 407 ± 27 | 395 ± 26 | 411 ± 26 |
| WT littermate mice | ||||||
| CON | 403 ± 14 | 414 ± 14 | 432 ± 15 | 421 ± 12 | 412 ± 15 | 408 ± 16 |
| l-NAME | 391 ± 11 | 389 ± 12 | 413 ± 15 | 401 ± 12 | 398 ± 12 | 390 ± 11 |
| IPC | 418 ± 20 | 408 ± 16 | 418 ± 15 | 419 ± 15 | 402 ± 18 | 396 ± 16 |
| IPC + l-NAME | 413 ± 11 | 393 ± 12 | 404 ± 12 | 406 ± 13 | 412 ± 13 | 419 ± 13 |
| IPC+HG | 381 ± 13 | 390 ± 13 | 402 ± 12 | 410 ± 15 | 415 ± 13 | 399 ± 17 |
| IPC+HG+l-NAME | 399 ± 12 | 383 ± 12 | 384 ± 9 | 374 ± 10 | 377 ± 14 | 390 ± 13 |
| Transgenic GTP cyclohydrolase 1 mice | ||||||
| CON | 391 ± 12 | 402 ± 12 | 415 ± 14 | 432 ± 15 | 427 ± 19 | 428 ± 20 |
| l-NAME | 395 ± 10 | 409 ± 19 | 412 ± 17 | 42- ± 16 | 413 ± 15 | 412 ± 17 |
| IPC | 393 ± 11 | 394 ± 13 | 405 ± 12 | 414 ± 14 | 391 ± 14 | 385 ± 15 |
| IPC + l-NAME | 393 ± 15 | 403 ± 11 | 401 ± 12 | 409 ± 15 | 419 ± 20 | 417 ± 16 |
| IPC + HG | 379 ± 22 | 424 ± 12 | 439 ± 17 | 431 ± 13 | 397 ± 18 | 398 ± 16 |
| IPC + HG + l-NAME | 409 ± 11 | 387 ± 17 | 418 ± 9 | 421 ± 10 | 414 ± 10 | 421 ± 10 |
| Group | Baseline | Coronary occlusion | Reperfusion | |||
|---|---|---|---|---|---|---|
| 2 min | 30 min | 60 min | 120 min | |||
| C57BL/6 mice | ||||||
| CON | 404 ± 12 | 411 ± 16 | 425 ± 18 | 413 ± 18 | 401 ± 21 | 391 ± 16 |
| HG | 393 ± 16 | 403 ± 14 | 399 ± 13 | 417 ± 15 | 404 ± 25 | 423 ± 13 |
| IPC | 418 ± 16 | 403 ± 14 | 414 ± 13 | 435 ± 12 | 394 ± 20 | 382 ± 15 |
| IPC+HG | 389 ± 26 | 412 ± 16 | 420 ± 12 | 408 ± 11 | 399 ± 16 | 408 ± 18 |
| IPC + HOsm | 422 ± 18 | 397 ± 19 | 401 ± 21 | 407 ± 27 | 395 ± 26 | 411 ± 26 |
| WT littermate mice | ||||||
| CON | 403 ± 14 | 414 ± 14 | 432 ± 15 | 421 ± 12 | 412 ± 15 | 408 ± 16 |
| l-NAME | 391 ± 11 | 389 ± 12 | 413 ± 15 | 401 ± 12 | 398 ± 12 | 390 ± 11 |
| IPC | 418 ± 20 | 408 ± 16 | 418 ± 15 | 419 ± 15 | 402 ± 18 | 396 ± 16 |
| IPC + l-NAME | 413 ± 11 | 393 ± 12 | 404 ± 12 | 406 ± 13 | 412 ± 13 | 419 ± 13 |
| IPC+HG | 381 ± 13 | 390 ± 13 | 402 ± 12 | 410 ± 15 | 415 ± 13 | 399 ± 17 |
| IPC+HG+l-NAME | 399 ± 12 | 383 ± 12 | 384 ± 9 | 374 ± 10 | 377 ± 14 | 390 ± 13 |
| Transgenic GTP cyclohydrolase 1 mice | ||||||
| CON | 391 ± 12 | 402 ± 12 | 415 ± 14 | 432 ± 15 | 427 ± 19 | 428 ± 20 |
| l-NAME | 395 ± 10 | 409 ± 19 | 412 ± 17 | 42- ± 16 | 413 ± 15 | 412 ± 17 |
| IPC | 393 ± 11 | 394 ± 13 | 405 ± 12 | 414 ± 14 | 391 ± 14 | 385 ± 15 |
| IPC + l-NAME | 393 ± 15 | 403 ± 11 | 401 ± 12 | 409 ± 15 | 419 ± 20 | 417 ± 16 |
| IPC + HG | 379 ± 22 | 424 ± 12 | 439 ± 17 | 431 ± 13 | 397 ± 18 | 398 ± 16 |
| IPC + HG + l-NAME | 409 ± 11 | 387 ± 17 | 418 ± 9 | 421 ± 10 | 414 ± 10 | 421 ± 10 |
All animals were subjected to 30 min of coronary occlusion followed by 2 h of reperfusion [control (CON)]. IPC was induced by four cycles of 5 min of coronary occlusion/5 min of reperfusion in the presence or absence of HG or hyperosmolarity (HOsm), as described in Figure 1A and B. There were no statistically significant difference among groups (n = 6–9 mice/group).
Heart rate (bpm) during in vivo study
| Group | Baseline | Coronary occlusion | Reperfusion | |||
|---|---|---|---|---|---|---|
| 2 min | 30 min | 60 min | 120 min | |||
| C57BL/6 mice | ||||||
| CON | 404 ± 12 | 411 ± 16 | 425 ± 18 | 413 ± 18 | 401 ± 21 | 391 ± 16 |
| HG | 393 ± 16 | 403 ± 14 | 399 ± 13 | 417 ± 15 | 404 ± 25 | 423 ± 13 |
| IPC | 418 ± 16 | 403 ± 14 | 414 ± 13 | 435 ± 12 | 394 ± 20 | 382 ± 15 |
| IPC+HG | 389 ± 26 | 412 ± 16 | 420 ± 12 | 408 ± 11 | 399 ± 16 | 408 ± 18 |
| IPC + HOsm | 422 ± 18 | 397 ± 19 | 401 ± 21 | 407 ± 27 | 395 ± 26 | 411 ± 26 |
| WT littermate mice | ||||||
| CON | 403 ± 14 | 414 ± 14 | 432 ± 15 | 421 ± 12 | 412 ± 15 | 408 ± 16 |
| l-NAME | 391 ± 11 | 389 ± 12 | 413 ± 15 | 401 ± 12 | 398 ± 12 | 390 ± 11 |
| IPC | 418 ± 20 | 408 ± 16 | 418 ± 15 | 419 ± 15 | 402 ± 18 | 396 ± 16 |
| IPC + l-NAME | 413 ± 11 | 393 ± 12 | 404 ± 12 | 406 ± 13 | 412 ± 13 | 419 ± 13 |
| IPC+HG | 381 ± 13 | 390 ± 13 | 402 ± 12 | 410 ± 15 | 415 ± 13 | 399 ± 17 |
| IPC+HG+l-NAME | 399 ± 12 | 383 ± 12 | 384 ± 9 | 374 ± 10 | 377 ± 14 | 390 ± 13 |
| Transgenic GTP cyclohydrolase 1 mice | ||||||
| CON | 391 ± 12 | 402 ± 12 | 415 ± 14 | 432 ± 15 | 427 ± 19 | 428 ± 20 |
| l-NAME | 395 ± 10 | 409 ± 19 | 412 ± 17 | 42- ± 16 | 413 ± 15 | 412 ± 17 |
| IPC | 393 ± 11 | 394 ± 13 | 405 ± 12 | 414 ± 14 | 391 ± 14 | 385 ± 15 |
| IPC + l-NAME | 393 ± 15 | 403 ± 11 | 401 ± 12 | 409 ± 15 | 419 ± 20 | 417 ± 16 |
| IPC + HG | 379 ± 22 | 424 ± 12 | 439 ± 17 | 431 ± 13 | 397 ± 18 | 398 ± 16 |
| IPC + HG + l-NAME | 409 ± 11 | 387 ± 17 | 418 ± 9 | 421 ± 10 | 414 ± 10 | 421 ± 10 |
| Group | Baseline | Coronary occlusion | Reperfusion | |||
|---|---|---|---|---|---|---|
| 2 min | 30 min | 60 min | 120 min | |||
| C57BL/6 mice | ||||||
| CON | 404 ± 12 | 411 ± 16 | 425 ± 18 | 413 ± 18 | 401 ± 21 | 391 ± 16 |
| HG | 393 ± 16 | 403 ± 14 | 399 ± 13 | 417 ± 15 | 404 ± 25 | 423 ± 13 |
| IPC | 418 ± 16 | 403 ± 14 | 414 ± 13 | 435 ± 12 | 394 ± 20 | 382 ± 15 |
| IPC+HG | 389 ± 26 | 412 ± 16 | 420 ± 12 | 408 ± 11 | 399 ± 16 | 408 ± 18 |
| IPC + HOsm | 422 ± 18 | 397 ± 19 | 401 ± 21 | 407 ± 27 | 395 ± 26 | 411 ± 26 |
| WT littermate mice | ||||||
| CON | 403 ± 14 | 414 ± 14 | 432 ± 15 | 421 ± 12 | 412 ± 15 | 408 ± 16 |
| l-NAME | 391 ± 11 | 389 ± 12 | 413 ± 15 | 401 ± 12 | 398 ± 12 | 390 ± 11 |
| IPC | 418 ± 20 | 408 ± 16 | 418 ± 15 | 419 ± 15 | 402 ± 18 | 396 ± 16 |
| IPC + l-NAME | 413 ± 11 | 393 ± 12 | 404 ± 12 | 406 ± 13 | 412 ± 13 | 419 ± 13 |
| IPC+HG | 381 ± 13 | 390 ± 13 | 402 ± 12 | 410 ± 15 | 415 ± 13 | 399 ± 17 |
| IPC+HG+l-NAME | 399 ± 12 | 383 ± 12 | 384 ± 9 | 374 ± 10 | 377 ± 14 | 390 ± 13 |
| Transgenic GTP cyclohydrolase 1 mice | ||||||
| CON | 391 ± 12 | 402 ± 12 | 415 ± 14 | 432 ± 15 | 427 ± 19 | 428 ± 20 |
| l-NAME | 395 ± 10 | 409 ± 19 | 412 ± 17 | 42- ± 16 | 413 ± 15 | 412 ± 17 |
| IPC | 393 ± 11 | 394 ± 13 | 405 ± 12 | 414 ± 14 | 391 ± 14 | 385 ± 15 |
| IPC + l-NAME | 393 ± 15 | 403 ± 11 | 401 ± 12 | 409 ± 15 | 419 ± 20 | 417 ± 16 |
| IPC + HG | 379 ± 22 | 424 ± 12 | 439 ± 17 | 431 ± 13 | 397 ± 18 | 398 ± 16 |
| IPC + HG + l-NAME | 409 ± 11 | 387 ± 17 | 418 ± 9 | 421 ± 10 | 414 ± 10 | 421 ± 10 |
All animals were subjected to 30 min of coronary occlusion followed by 2 h of reperfusion [control (CON)]. IPC was induced by four cycles of 5 min of coronary occlusion/5 min of reperfusion in the presence or absence of HG or hyperosmolarity (HOsm), as described in Figure 1A and B. There were no statistically significant difference among groups (n = 6–9 mice/group).
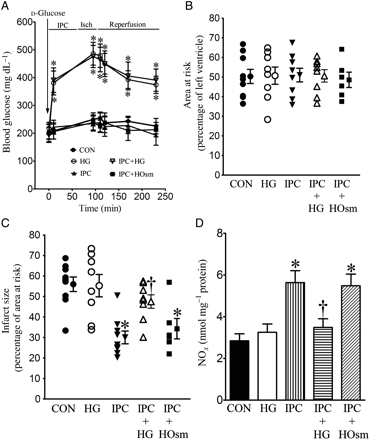
HG inhibited decreases in myocardial infarct size produced by IPC in WT mice subjected to I/R injury. (A) Blood glucose concentrations during myocardial ischaemia (Isch) and reperfusion; (B) AAR expressed as the percentage of the left ventricle; (C) infarct size expressed as the percentage of AAR. All mice were subjected to 30 min of coronary occlusion followed by 2 h of reperfusion (CON). IPC was induced by four cycles of 5 min of ischaemia/5 min of reperfusion. Mice were injected with d-glucose to produce HG or mannitol to produce hyperosmolarity (HOsm); (D) NO concentrations. The hearts were collected 5 min after reperfusion, and myocardial NO and its metabolites (nitrate and nitrite) were measured. *P < 0.05 vs. CON, †P < 0.05 vs. IPC (n = 6–10 mice/group).
IPC significantly increased NO production in myocardium (Figure 2D) measured after 30 min of coronary occlusion and 5 min of reperfusion compared with control hearts that were not preconditioned. HG had no effect on NO production in the absence of IPC, but reduced increases in NO produced by IPC. In contrast to the effects of HG, mannitol did not attenuate increases in NO production during IPC.
3.2 Cardiomyocyte-targeted overexpression of the human GTPCH-1 gene increased myocardial BH4 content
Expression of human GTPCH-1 mRNA normalized to GAPDH was significantly increased in hearts of GTPCH-Tg mice compared with WT littermates (Figure 3; n = 5, P < 0.05). Overexpression of the human GTPCH-1 gene did not alter expression of mouse GTPCH-1 mRNA. Myocardial BH4 content was dramatically increased in GTPCH-Tg hearts (n = 5) compared with WT littermates.
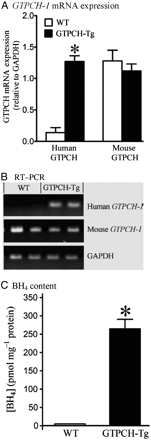
Cardiomyocyte-specific overexpression of the human GTP cyclohydrolase (GTPCH) 1 gene increased myocardial BH4 content. (A) Densitometry normalized to the GAPDH house-keeping gene showing expression of human and mouse GTPCH mRNA in hearts of transgenic (GTPCH-Tg) vs. WT littermate (WT) mice (human GTPCH mRNA was absent in WT mice and the histogram represents background densitometry values); (B) RT–PCR showing expression of human and mouse GTPCH mRNA and GAPDH in hearts of GTPCH-Tg mice vs. WT littermates; (C) BH4 levels in GTPCH-Tg and WT littermate hearts. Student's t-test was used to analyse the difference between GCTPH-Tg and WT littermate groups. *P < 0.05 vs. WT (n = 5 hearts/group).
3.3 GTPCH-1 overexpression rescued IPC during HG via a NO-dependent mechanism
There were no significant differences in the heart rate at baseline, or during coronary occlusion and reperfusion (Table 1) in WT littermate and transgenic mice. AAR was comparable between groups. Myocardial infarct size (Figure 4 and Supplementary material online, Figure S1) was significantly decreased by IPC in WT littermates compared with control mice. IPC produced a similar degree of protection against infarction in WT and GTPCH-Tg mice. Decreases in infarct size produced by IPC alone were not altered by l-NAME in either WT or GTPCH-Tg mice. HG inhibited the protective effect of IPC in WT littermates but not in GTPCH-Tg mice. Interestingly, l-NAME blocked the beneficial effects of GTPCH-1 overexpression during IPC and HG.
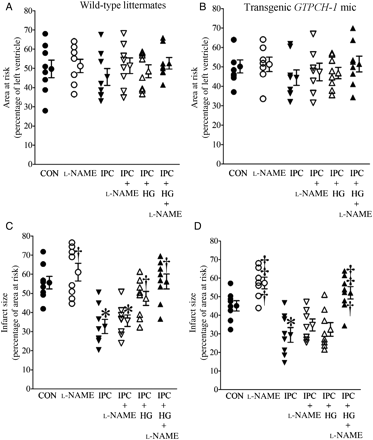
Overexpression of the GTP cyclohydrolase (GTPCH) 1 gene rescued cardioprotection produced by IPC in the presence of HG through a NO-dependent mechanism. (A and B) AAR expressed as a percentage of left ventricle; (C and D) infarct size expressed as a percentage of AAR. Control (CON) mice were subjected to 30 min of coronary occlusion followed by 2 h of reperfusion. IPC was elicited with four cycles of 5 min of ischaemia/5 min of reperfusion. l-NAME (1 mg/kg) was administered intraperitoneally 80 min before coronary artery occlusion or 40 min before IPC. *P < 0.05 vs. CON; †P < 0.05 vs. IPC; ‡P < 0.05 vs. IPC + HG (n = 7–8 mice/group).
Echocardiography was used to examine whether cardiomyocyte-specific overexpression of GTPCH-1 altered baseline cardiac function. There were no significant differences in multiple cardiac functional indices (Table 2), including heart rate; LV diameter in diastole and systole; fractional shortening; peak velocity of mitral E and A waves; mitral E/A ratio; isovolumic contraction time; ejection time; isovolumic relaxation time; myocardial performance index; mitral E acceleration; E wave acceleration time; mitral E deceleration; and E wave deceleration time between C57BL/6, GTPCH-Tg, and WT littermate mice. Only LV wall thickness was found to be significantly increased in GTPCH-Tg mice compared with WT mice.
Echocardiographic parameters in C57BL/6, transgenic GTP chclohydrolase 1 (GTPCH-Tg), and WT littermate mice
| C57BL/6 | Littermates | GTPCH-Tg | |
|---|---|---|---|
| Heart rate (bpm) | 456 ± 22 | 442 ± 26 | 461 ± 15 |
| AWd (mm) | 0.78 ± 0.04 | 0.82 ± 0.05 | 0.98 ± 0.05* |
| AWs (mm) | 1.24 ± 0.10 | 1.25 ± 0.08 | 1.44 ± 0.07* |
| PWd (mm) | 0.81 ± 0.07 | 0.83 ± 0.07 | 1.01 ± 0.05* |
| PWs (mm) | 1.23 ± 0.12 | 1.21 ± 0.07 | 1.36 ± 0.03* |
| LVIDd (mm) | 3.78 ± 0.12 | 3.82 ± 0.18 | 3.93 ± 0.11 |
| LVIDs (mm) | 2.30 ± 0.15 | 2.49 ± 0.17 | 2.51 ± 0.09 |
| Fractional shortening (%) | 39 ± 3 | 35 ± 3 | 36 ± 2 |
| Peak E (cm/s) | 82 ± 4 | 83 ± 4 | 78 ± 5 |
| Peak A (cm/s) | 50 ± 4 | 48 ± 5 | 43 ± 3 |
| Mitral E/A ratio | 1.68 ± 0.09 | 1.80 ± 0.13 | 1.83 ± 0.13 |
| IVCT (ms) | 12.6 ± 0.77 | 13.2 ± 0.8 | 14.8 ± 0.6 |
| Ejection time (ms) | 42.8 ± 1.2 | 43.0 ± 1.3 | 44.4 ± 1.3 |
| IVRT (ms) | 16.0 ± 0.4 | 16.6 ± 0.9 | 16.4 ± 0.7 |
| MPI | 0.67 ± 0.02 | 0.69 ± 0.03 | 0.71 ± 0.03 |
| Mitral E acceleration (cm/ms) | 8001 ± 657 | 8361 ± 785 | 8058 ± 681 |
| Eat (ms) | 9.2 ± 0.6 | 9.0 ± 0.7 | 9.7 ± 0.6 |
| Mitral E deceleration (cm/ms) | 4476 ± 596 | 4439 ± 548 | 4187 ± 428 |
| Edt (ms) | 16.7 ± 1.4 | 16.2 ± 1.1 | 17.6 ± 0.7 |
| C57BL/6 | Littermates | GTPCH-Tg | |
|---|---|---|---|
| Heart rate (bpm) | 456 ± 22 | 442 ± 26 | 461 ± 15 |
| AWd (mm) | 0.78 ± 0.04 | 0.82 ± 0.05 | 0.98 ± 0.05* |
| AWs (mm) | 1.24 ± 0.10 | 1.25 ± 0.08 | 1.44 ± 0.07* |
| PWd (mm) | 0.81 ± 0.07 | 0.83 ± 0.07 | 1.01 ± 0.05* |
| PWs (mm) | 1.23 ± 0.12 | 1.21 ± 0.07 | 1.36 ± 0.03* |
| LVIDd (mm) | 3.78 ± 0.12 | 3.82 ± 0.18 | 3.93 ± 0.11 |
| LVIDs (mm) | 2.30 ± 0.15 | 2.49 ± 0.17 | 2.51 ± 0.09 |
| Fractional shortening (%) | 39 ± 3 | 35 ± 3 | 36 ± 2 |
| Peak E (cm/s) | 82 ± 4 | 83 ± 4 | 78 ± 5 |
| Peak A (cm/s) | 50 ± 4 | 48 ± 5 | 43 ± 3 |
| Mitral E/A ratio | 1.68 ± 0.09 | 1.80 ± 0.13 | 1.83 ± 0.13 |
| IVCT (ms) | 12.6 ± 0.77 | 13.2 ± 0.8 | 14.8 ± 0.6 |
| Ejection time (ms) | 42.8 ± 1.2 | 43.0 ± 1.3 | 44.4 ± 1.3 |
| IVRT (ms) | 16.0 ± 0.4 | 16.6 ± 0.9 | 16.4 ± 0.7 |
| MPI | 0.67 ± 0.02 | 0.69 ± 0.03 | 0.71 ± 0.03 |
| Mitral E acceleration (cm/ms) | 8001 ± 657 | 8361 ± 785 | 8058 ± 681 |
| Eat (ms) | 9.2 ± 0.6 | 9.0 ± 0.7 | 9.7 ± 0.6 |
| Mitral E deceleration (cm/ms) | 4476 ± 596 | 4439 ± 548 | 4187 ± 428 |
| Edt (ms) | 16.7 ± 1.4 | 16.2 ± 1.1 | 17.6 ± 0.7 |
AWd, anterior wall (interventricular septum) at end-diastole; AWs, anterior wall at end systole; PWd, posterior wall at diastole; PWs, posterior wall at systole; LVIDd, LV internal diameter at diastole; LVIDs, LV internal diameter at systole; fractional shortening, 100 × (LVIDd-LVIDs)/LVIDd; Peak E, peak velocity of mitral E valve; Peak A, peak velocity of mitra A wave; IVCT, isovolumic contraction time of LV; IVRT, isovolumic relaxation time of LV; MPI (myocardial performance index) = the ratio of the sum of isovolumic contraction and relaxation times to ejection time; Eat, mitral E wave acceleration time; Edt, E wave deceleration time.
*P < 0.05 vs. C57BL/6 and WT littermates (n = 10 mice/group).
Echocardiographic parameters in C57BL/6, transgenic GTP chclohydrolase 1 (GTPCH-Tg), and WT littermate mice
| C57BL/6 | Littermates | GTPCH-Tg | |
|---|---|---|---|
| Heart rate (bpm) | 456 ± 22 | 442 ± 26 | 461 ± 15 |
| AWd (mm) | 0.78 ± 0.04 | 0.82 ± 0.05 | 0.98 ± 0.05* |
| AWs (mm) | 1.24 ± 0.10 | 1.25 ± 0.08 | 1.44 ± 0.07* |
| PWd (mm) | 0.81 ± 0.07 | 0.83 ± 0.07 | 1.01 ± 0.05* |
| PWs (mm) | 1.23 ± 0.12 | 1.21 ± 0.07 | 1.36 ± 0.03* |
| LVIDd (mm) | 3.78 ± 0.12 | 3.82 ± 0.18 | 3.93 ± 0.11 |
| LVIDs (mm) | 2.30 ± 0.15 | 2.49 ± 0.17 | 2.51 ± 0.09 |
| Fractional shortening (%) | 39 ± 3 | 35 ± 3 | 36 ± 2 |
| Peak E (cm/s) | 82 ± 4 | 83 ± 4 | 78 ± 5 |
| Peak A (cm/s) | 50 ± 4 | 48 ± 5 | 43 ± 3 |
| Mitral E/A ratio | 1.68 ± 0.09 | 1.80 ± 0.13 | 1.83 ± 0.13 |
| IVCT (ms) | 12.6 ± 0.77 | 13.2 ± 0.8 | 14.8 ± 0.6 |
| Ejection time (ms) | 42.8 ± 1.2 | 43.0 ± 1.3 | 44.4 ± 1.3 |
| IVRT (ms) | 16.0 ± 0.4 | 16.6 ± 0.9 | 16.4 ± 0.7 |
| MPI | 0.67 ± 0.02 | 0.69 ± 0.03 | 0.71 ± 0.03 |
| Mitral E acceleration (cm/ms) | 8001 ± 657 | 8361 ± 785 | 8058 ± 681 |
| Eat (ms) | 9.2 ± 0.6 | 9.0 ± 0.7 | 9.7 ± 0.6 |
| Mitral E deceleration (cm/ms) | 4476 ± 596 | 4439 ± 548 | 4187 ± 428 |
| Edt (ms) | 16.7 ± 1.4 | 16.2 ± 1.1 | 17.6 ± 0.7 |
| C57BL/6 | Littermates | GTPCH-Tg | |
|---|---|---|---|
| Heart rate (bpm) | 456 ± 22 | 442 ± 26 | 461 ± 15 |
| AWd (mm) | 0.78 ± 0.04 | 0.82 ± 0.05 | 0.98 ± 0.05* |
| AWs (mm) | 1.24 ± 0.10 | 1.25 ± 0.08 | 1.44 ± 0.07* |
| PWd (mm) | 0.81 ± 0.07 | 0.83 ± 0.07 | 1.01 ± 0.05* |
| PWs (mm) | 1.23 ± 0.12 | 1.21 ± 0.07 | 1.36 ± 0.03* |
| LVIDd (mm) | 3.78 ± 0.12 | 3.82 ± 0.18 | 3.93 ± 0.11 |
| LVIDs (mm) | 2.30 ± 0.15 | 2.49 ± 0.17 | 2.51 ± 0.09 |
| Fractional shortening (%) | 39 ± 3 | 35 ± 3 | 36 ± 2 |
| Peak E (cm/s) | 82 ± 4 | 83 ± 4 | 78 ± 5 |
| Peak A (cm/s) | 50 ± 4 | 48 ± 5 | 43 ± 3 |
| Mitral E/A ratio | 1.68 ± 0.09 | 1.80 ± 0.13 | 1.83 ± 0.13 |
| IVCT (ms) | 12.6 ± 0.77 | 13.2 ± 0.8 | 14.8 ± 0.6 |
| Ejection time (ms) | 42.8 ± 1.2 | 43.0 ± 1.3 | 44.4 ± 1.3 |
| IVRT (ms) | 16.0 ± 0.4 | 16.6 ± 0.9 | 16.4 ± 0.7 |
| MPI | 0.67 ± 0.02 | 0.69 ± 0.03 | 0.71 ± 0.03 |
| Mitral E acceleration (cm/ms) | 8001 ± 657 | 8361 ± 785 | 8058 ± 681 |
| Eat (ms) | 9.2 ± 0.6 | 9.0 ± 0.7 | 9.7 ± 0.6 |
| Mitral E deceleration (cm/ms) | 4476 ± 596 | 4439 ± 548 | 4187 ± 428 |
| Edt (ms) | 16.7 ± 1.4 | 16.2 ± 1.1 | 17.6 ± 0.7 |
AWd, anterior wall (interventricular septum) at end-diastole; AWs, anterior wall at end systole; PWd, posterior wall at diastole; PWs, posterior wall at systole; LVIDd, LV internal diameter at diastole; LVIDs, LV internal diameter at systole; fractional shortening, 100 × (LVIDd-LVIDs)/LVIDd; Peak E, peak velocity of mitral E valve; Peak A, peak velocity of mitra A wave; IVCT, isovolumic contraction time of LV; IVRT, isovolumic relaxation time of LV; MPI (myocardial performance index) = the ratio of the sum of isovolumic contraction and relaxation times to ejection time; Eat, mitral E wave acceleration time; Edt, E wave deceleration time.
*P < 0.05 vs. C57BL/6 and WT littermates (n = 10 mice/group).
3.4 HG decreased cardiac BH4 and NO concentrations during IPC
Baseline cardiac BH4 concentrations were significantly higher in GTPCH-Tg hearts than in WT hearts (Figure 5A and B). IPC increased BH4 concentrations in WT but not GTPCH-Tg hearts. HG decreased BH4 concentrations in GTPCH-Tg hearts and in both WT and GTPCH-Tg hearts during IPC. BH4 concentrations were significantly higher in GTPCH-Tg hearts than in WT hearts during IPC in the presence of HG (n = 5, P < 0.05).
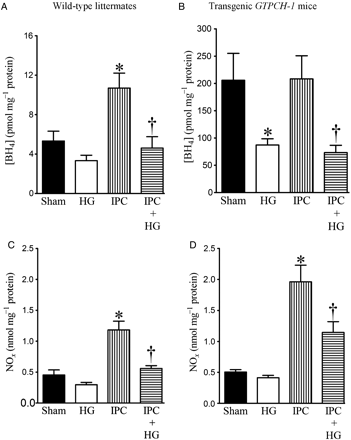
Effects of HG on myocardial tetrahydrobiopterin (BH4) and nitric oxide (NOx) concentrations in WT littermates and transgenic GTPCH-1 mice during IPC. (A and B) Myocardial BH4 concentrations in WT and transgenic GTPCH-1 mice, respectively; (C and D) NOx production in WT and transgenic GTPCH-1 mice, respectively. WT littermates and transgenic GTPCH-1 mice underwent a sham operation (sham) or IPC induced by four cycles of 5 min of ischaemia/5 min of reperfusion in the presence or absence of HG. Mice were not subjected to prolonged coronary artery occlusion and reperfusion. The hearts were excised 5 min after the final IPC and immediately frozen for measurement of BH4 or NOx. *P < 0.05 vs. sham; †P < 0.05 vs. IPC (n = 5–9 mice/group).
There were no significant differences in baseline NOx concentrations between GTPCH-Tg and WT hearts (Figure 5C and D). HG did not change baseline NOx concentrations but decreased IPC-elicited increases in NOx production. IPC-elicited increases in NOx production were significantly higher in GTPCH-Tg hearts than in WT hearts in the presence and absence of HG (n = 9, P < 0.05).
3.5 HG reduced IPC-elicited protection against mPTP opening in WT littermates but not GTPCH-Tg hearts
In the WT sham group, the concentration of in vitro Ca2+ loading necessary to open the mPTP was 253 ± 5 µmol CaCl2 mg−1 protein (n = 10). This concentration was reduced (P < 0.05) in mitochondria isolated from WT hearts subjected to I/R (Figure 6). IPC significantly increased the amount of Ca2+ required to elicit mPTP opening, indicating protection. HG abrogated the protective effect of IPC in WT littermates and increased the sensitivity of mPTP opening to CaCl2. In contrast, HG did not attenuate the protective effects of IPC on mPTP opening in GTPCH-Tg mice. l-NAME did not change the effects of I/R and IPC on mPTP in both WT and GTPCH-Tg mice; however, l-NAME blocked the beneficial effect of IPC on mPTP in hyperglycaemic, GTPCH-Tg mice.
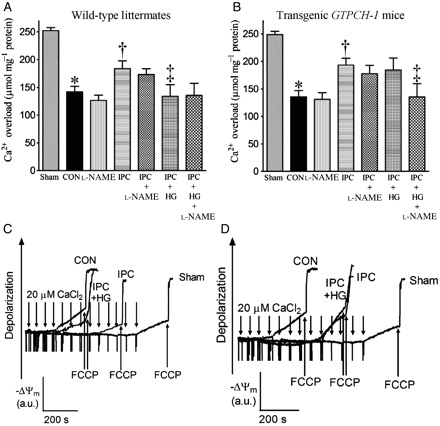
Effects of HG or l-NAME on opening of the mPTP in WT littermates and transgenic GTPCH-1 mice with and without IPC in vivo. (A and B) The amount of in vitro Ca2+ overload required to open the mPTP pore in WT littermate and transgenic GTPCH-1 mice, respectively; (C and D) representative tracings showing the changes in membrane potential (ΔΨm) of mitochondria isolated from WT littermate or transgenic GTPCH-1 hearts after in vitro Ca2+ loading, respectively. Opening of the mPTP pore was assessed by following changes in ΔΨm using the fluorescent dye rhodamine 123. Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) was added at the arrows to depolarize mitochondria. a.u., arbitrary unit. *P < 0.05 vs. sham; †P < 0.05 vs. CON; ‡P < 0.05 vs. IPC (n = 9–10/group).
4. Discussion
The results of the present investigation confirm and extend previous findings that HG reduce the beneficial effects of IPC to protect against myocardial infarction and inhibit mPTP opening.3–6 The results further indicate that BH4 and NO assume an important role during IPC in the presence of HG. Interestingly, during HG myocardial overexpression of GTPCH-1 increases BH4 and NO concentrations, restores the efficacy of IPC, and these actions are blocked by NOS inhibition.
BH4 is synthesized in the cell cytoplasm by either de novo or salvage pathways,19 and the activity of GTPCH-1 is critical for the production of BH4 by the de novo pathway. The intracellular regulation of BH4 is dependent on the cellular redox state. Acute HG has previously been shown to cause intracellular BH4 deficiency by increasing oxidative stress.20 For example, ONOO– is increased by HG and this oxidant species oxidizes BH4 to the catalytically incompetent species dihydrobiopterin.21 Evidence indicates that high concentrations of glucose decrease BH4 concentrations concomitantly with reductions in NO production and increases in O2•− anion.22,23 This action may occur as a result of uncoupling of the NOS enzyme. NOS enzyme consists of a haem-containing oxygenase domain that binds BH4, molecular oxygen, and l-arginine; and a reductase domain that transfers electrons from reduced NADP to FAD and FMN.24 In the presence of adequate substrate, l-arginine, and cofactor BH4, haem and oxygen reduction are coupled to the synthesis of NO. However, electron transfer within the active site of NOS can become uncoupled from l-arginine oxidation during conditions of low intracellular BH4. Thus, NOS uncoupling causes molecular oxygen to be reduced to O2•−.25 Interestingly, O2•− is the sole product of recombinant eNOS in the absence of BH4 and conversely, increases in BH4 favour enhanced production of NO, a decrease in O2•−,26 and maintenance of eNOS in a phosphorylated active form.20
NOS-derived NO has been shown to produce cardioprotection27 and to be a crucial mediator of the late phase of IPC.28 However, the role of NOS/NO in the early phase of IPC is controversial.29–32 In the present study, l-NAME did not block the early phase of IPC to protect against myocardial infarction in normoglycemic WT mice, in agreement with a recent study by Guo et al. in mice.32 Brief episodes of preconditioning ischaemia and reperfusion cause the release of adenosine, opioids, bradykinin, and oxygen radicals in addition to NO, and these mediators act on multiple signalling pathways to elicit myocardial protection.33 It is possible that redundant signalling mechanisms can compensate for a loss of NO, and thus, NO is not required to trigger protection in normal myocardium. However, l-NAME blocked the beneficial effects of GTPCH-1 overexpression during IPC and HG. HG has been demonstrated to adversely affect multiple pathways of cardioprotective signalling.34,35 Unlike normal myocardium, other preconditioning mediators may fail to compensate for loss of NO in hyperglycaemic myocardium. Interestingly, NO levels in GTPCH-Tg hearts during IPC and HG were maintained at levels roughly equal to the WT hearts with IPC alone. Augmented BH4/NO signalling by cardiomyocyte-specific overexpression of GTPCH-1 appeared to overcome the signal transduction defects induced by HG.
We have previously demonstrated that diabetes and HG block infarct size reduction in response to ischaemic and pharmacological preconditioning.3,6,36,37 and that sepiapterin, a metabolic precursor of BH4, restores NO production during HG.23 The current results provide critical evidence that genetic strategies designed to increase BH4 bioavailability during HG also enhance cardioprotection. This contention is supported by other evidence that myocardial I/R injury decreases intracellular BH4 concentrations in parallel with overproduction of O2•−,38 while in contrast, supplementation of BH4 improves LV function after I/R.39 Exogenous BH4 also increases NO production and decreases O2•− concentrations during conditions, such as diabetes,40 acute HG,41 hypercholesterolaemia,42 and chronic smoking.11 Interestingly, we have demonstrated that the 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitor simvastatin restores IPC during HG through a NO-dependent mechanism6 and statins have been shown to stimulate the synthesis of BH4 by increasing the expression of GTPCH-1.43 Taken together, the results indicate that targeting BH4 directly with cardiomyocyte-specific overexpression of the GTPCH-1 gene, or indirectly with statins, may represent important therapeutic approaches to restoring cardioprotective signalling during diabetes and HG. Endothelium-targeted overexpression of GTPCH-1 by gene transfer has been reported to maintain endothelial BH4 content during HG and diabetes and to restore both the activity of eNOS and endothelial-dependent relaxation.9,20 Our novel results indicate that cardiomyocyte-specific overexpression of this gene restores IPC during relative eNOS deficiency as observed with acute HG. In addition, overexpression of the GTPCH-1 gene and subsequently preserved BH4 concentrations during HG may have maintained NOS in a coupled state; however, this hypothesis remains to be specifically tested.
The current findings also indicate that during HG GTPCH-1 overexpression protects mitochondria against I/R injury. The importance of mitochondria as both targets and mediators of I/R injury is becoming increasingly recognized.44 mPTP is an oxidative stress-sensitive channel involved in cell death.45 It remains closed during myocardial ischaemia but opens during the first several minutes of reperfusion due to Ca2+ overload and excessive production of reactive oxygen species.45 The opening of the mPTP results in depolarization of the membrane potential and matrix swelling, which leads to rupture of the outer membrane and release of proteins such as cytochrome c from the intermembrane space into the cytosol. HG produces excessive amounts of O2•− by mitochondria and favours mPTP opening and subsequent cell death.46 IPC has previously been demonstrated to inhibit mPTP opening at reperfusion and this action appears to be a key downstream effector of IPC.47 Although not directly established, the current results support the contention that HG may have enhanced mPTP opening, whereas overexpression of the GTPCH-1 gene restored the ability of IPC to prevent mPTP opening during HG.
The current results should be interpreted within the constraints of several potential limitations. Mannitol used as an osmolarity control is a reactive oxygen species scavenger and could contribute to cardioprotection. However, we have previously demonstrated that the non-metabolic sugar raffinose that does not scavenger reactive oxygen species fails to block IPC, in contrast to the findings during administration of glucose.48 In addition, BH4 is an essential co-factor for several other enzymes, including tyrosine hydroxylase, phenylalanine hydroxylase, and tryptophan hydroxylase, and modulates the synthesis of catecholamines. It is unknown whether cardiomyocyte-specific overexpression of the GTPCH-1 gene might have increased cardiac catecholamine synthesis. However, there were no changes in heart rate or echocardiographic indices of ventricular function to suggest that BH4-induced changes in basal catecholamine levels played a significant role in the results observed during this investigation. The results show that BH4 levels were substantially higher in GTPCH-Tg compared with WT mice at baseline and BH4 was not further increased by IPC. However, IPC significantly enhanced NO production during HG in GTPCH-Tg mice and NO levels were similar to that observed during IPC in WT mice. Thus, enhancing BH4 may preserve the fidelity of NO production in response to IPC stimuli despite HG. Although l-NAME blocked the productive effect of GTPCH-1 overexpression, we did not directly examine the effects of l-NAME on NO production in the mice.
In summary, the findings demonstrate that IPC failed to attenuate mPTP opening during HG. In contrast, cardiomyocyte-specific overexpression of the GTPCH-1 gene restored the efficacy of IPC to decrease myocardial I/R injury during HG by increasing bioavailability of BH4 and NO, and this was blocked by NOS inhibition. Thus, genetic modulation of GTPCH-1 may represent a novel approach to the treatment of cardiovascular disease during diabetes and HG.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported, in part, by National Institutes of Health research grants HL 063705 (to J.R.K.), HL 079837 (to G.M.P.), HL 054820 (to D.C.W.), and GM 066730 (to J.R.K. and D.C.W.) from the United States Public Health Services, Bethesda, Maryland.
Acknowledgements
We thank Dr Garrett J. Gross (Professor of Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, USA) for critical reading of the manuscript, Mark R. Paterson, John G. Krolikowski, David Schwabe, Shelley L. Baumgardt (all from Department of Anesthesiology, Medical College of Wisconsin), and Jennifer J. Whitselt (from Department of Biophysics, Medical College of Wisconsin) for excellent technical assistance.
Conflict of interest: this manuscript was presented in part at the 2010 American Heart Association Scientific Sessions, Chicago, Illinois, USA, 13–17 November 2010 and published in abstract form (Circulation 2010;122:A17272).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}