





Many cell culture and animal studies suggest that an increased formation of reactive oxygen species (ROS) promotes smooth muscle cell proliferation and matrix formation. It is therefore believed that increased ROS production contributes to such different entities as hypertension, restenosis, or arteriosclerosis. Initially, redox processes were believed to be mediated predominantly by hydrogen peroxide (H2O2). Over the years, however, many new aspects of redox signalling have been identified such as delicate alterations in the thiol redox state affecting the cellular redox potential, the complex interplay of nitric oxide with ROS, including the subsequent formation of peroxynitrite, and finally the involvement of peroxidases in redox signalling.1
Peroxidases catalyse the oxidation of a broad spectrum of substrates by H2O2, which, in the case of thyroid peroxidase, is needed for the oxidation of iodine and the formation of thyroid hormone (TPO), or in the case of myeloperoxidase (MPO) results in the formation of hypochlorous acid (HOCl), an important mediator of host defense.2 In the cardiovascular system, MPO contributes to vascular dysfunction; it promotes lipid peroxidation, scavenges nitric oxide and MPO products activate matrix metalloproteinases, promoting tissue remodelling that can result in atrial fibrillation.3
In addition to TPO and MPO, lactoperoxidase, eosinophil peroxidase, and salivary peroxidase belong to the family of animal peroxidases. Recently, vascular peroxidase (VPO1), also known as mammalian homologue of peroxidasin, was added to this list,4 although the physiological function of VPO1 remains unknown.
In this issue of Cardiovascular Research, Shi et al.5 provide compelling evidence that VPO1 contributes to angiotensin II (AngII)-induced proliferation of vascular smooth muscle cells (SMC). VPO1 protein expression was readily detectable in the A10 smooth muscle cell line and down-regulation of the protein attenuated AngII-stimulated proliferation. The mRNA and protein of VPO1 were induced by AngII, and VPO1 expression was also higher in vessels of spontaneously hypertensive rats when compared with Wistar Kytoto control rats. These data identify VPO1 as a novel mediator of AngII signalling. Interestingly, VPO1 expression was at least in part mediated by NADPH oxidase activation, and, thus, H2O2 itself could also induce the enzyme. Given that VPO1 utilizes NADPH oxidase-derived H2O2 to form peroxides,4 it is attractive to speculate that the system is a novel mediator of AngII-stimulated peroxide formation. Indeed, Shi et al. demonstrate increased formation of HOCl after AngII stimulation, and HOCl production was sensitive in part to VPO1 down-regulation by siRNA. Accordingly, H2O2-induced HOCl formation of A10 SMCs was also attenuated by VPO1 siRNA.
These observations demonstrate that VPO1 is a functionally important peroxidase in the vascular system and that VPO1 is a mediator of AngII-induced proliferation. A central aspect, however, remains open in this study: is the pro-proliferative effect of VPO1 also mediated by the HOCl formation of the enzyme? The authors provide no data to suggest that down-regulation of VPO1 affects tissue chloro-tyrosine adduct levels, a footprint marker of the biological activity of HOCl, and they also did not attempt to pharmacologically inhibit VPO1, thus blocking HOCl formation without affecting protein production. Peroxidase inhibitors that would also block VPO1 activity, like 4-aminobenzoic acid hydrazide, are available and bio-compatible. Surprisingly, in cholesterol-fed rabbits, peroxidase inhibition enhanced rather than reduced vascular hypertrophy and arteriosclerosis.6 Although peroxidases, by several rather negative mechanisms like uncoupling of endothelial nitric oxide synthase or oxidation of lipoproteins, should per se promote vascular disease,2 it is not known whether these effects can also be mediated by VPO1. Indeed, VPO appears to have a very low enzymatic activity compared with other peroxidases.4 In fact, convincing data have been presented to suggest that at least some of the effects of VPO1 are peroxide independent: Recently, Peterfi et al.7 characterized the function and expression of VPO1 in renal cells and observed that the protein is induced and secreted upon stimulation with transforming growth factor (TGF)-β1. Based on the homology to the insect peroxidase peroxidasin, the authors called VPO1 by that name and thus peroxidasin and VPO1 are identical. Importantly, in addition to being a peroxidase and being different from all other peroxidases, VPO1 carries several features of a matrix protein like a signalling peptide, a von Willebrand factor type C domain, leucine-rich repeats, and immunoglobulin G domains.8 Upon pro-fibrotic stress, as occurs in the unilateral ureteral obstruction model of the kidney, and in response to TGF β1 in cultured renal cells, large amounts of VPO1/peroxidasin are secreted into the extracellular space, where it organizes into fibrillar-like structures without any significant peroxidase activity.7
It is currently unknown whether the matrix function of VPO1/peroxidasin also contributes to the effects of AngII reported in the study by Shi et al.,5 but there are similar examples of such stimulatory functions of matrix proteins in the literature, such as biglycan. This matrix protein also contains leucine-rich repeats, stimulates smooth muscle cell proliferation and migration,9 and links matrix to inflammatory activation10 (Figure 1).
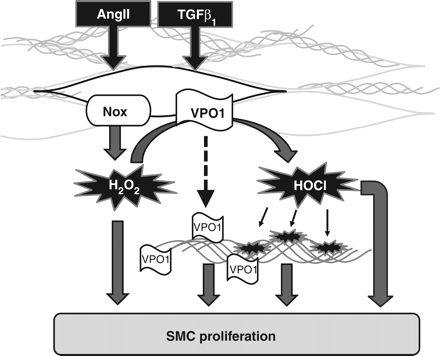
Possible pathways linking VPO1 to cell proliferation. AngII and TGFβ1 increase activity of Nox proteins and induce VPO1. For the sake of clarity, individual Nox (NADPH oxidase) proteins, although they differ in localization, mode of activation, and induction in response to AngII and TGFβ1 as well as type of ROS formed, are not differentiated in the illustration.13 VPO1 might subsequently utilize Nox-derived H2O2 to form HOCl, which modifies proteins. VPO1 could also be secreted to bind to matrix proteins and alter matrix signalling. Thus, VPO1, directly or indirectly through protein modifications or a direct action, might stimulate cell proliferation. These novel signalling aspects could amplify the proliferative response of smooth muscle cells to H2O2. The contribution of each pathway may vary between tissues and disease scenario.
It is remarkable that pro-fibrotic agents like AngII and TGF β1 stimulate peroxidase activity by a dual mode of action: by increasing the supply of substrate by inducing and activating NADPH oxidases and by simultaneously inducing the peroxidase itself. The biological consequence of this axis in chordata like mammals is completely elusive. In lower organisms like sea urchin and C. elegans, such a system is used to stabilize or seal extracellular matrix proteins by tyrosine crosslinking.11 In chordata, collagen crosslinking is mediated by the lysyl oxidase, a copper-containing enzyme that does not require H2O2 and even generates H2O2 itself as a by-product of lysine activation. Interestingly, lysyl oxidase is also induced by TGF β1.12 Nevertheless, it appears possible that even in mammals, matrix proteins are crosslinked by the NADPH oxidase–peroxidase axis, but such a matrix protein has not yet been identified.
Conflict of interest: none declared.
Funding
This manuscript was supported by grants from the Deutsche Forschungsgemeinschaft (SFB815/TP1 and SFB834/TPA2) and the excellence cluster cardio-pulmonary system (ECCPS).
References
Author notes
The opinions expressed in this article are not necessarily those of the Editors of Cardiovascular Research or of the European Society of Cardiology.

{kind=link}