





Abstract
Cigarette smoking engenders inflammation and endothelial dysfunction, processes implicated in atherothrombotic disease. We hypothesized that an interaction between inflammatory cytokines in smokers' blood and circulating components of cigarette smoke is necessary to induce reactive oxygen species (ROS) and cyclooxygenase-2 (COX-2) in endothelium. We then explored the molecular mechanisms involved in these effects.
Serum from nine healthy active smokers (AS) compared with serum from nine non-smokers (NS) showed higher levels of interleukin-1beta (IL-1β) and tumour necrosis factor-alpha (TNF-α) and a greater ability to induce ROS production, p47phox translocation to the plasma membrane, and COX-2 mRNA and protein expression in endothelial cells (ECs). Similar results were obtained in vivo and in vitro after treatment with aqueous extracts of cigarette smoke plus IL-1β and TNF-α(TS/IL-1β/TNF-α). In ECs increased ROS production and COX-2 mRNA induced by serum from AS correlated positively with their serum levels of IL-1β and TNF-α. Moreover, a positive correlation was observed between ROS generation and COX-2 mRNA. Simultaneous immuno-neutralization of IL-1β and TNF-α prevented endothelial dysfunction induced by serum from AS. Inhibitors of NADPH oxidase and/or p47phox siRNA diminished ROS production and COX-2 expression as well as phosphorylation of p38 mitogen-activated protein kinase (p38MAPK) and Akt mediated either by AS serum or by TS/IL-1β/TNF-α. Finally, direct inhibition of p38MAPK and Akt activity also abolished COX-2 expression mediated by both types of stimuli. Our results suggest a crucial role played by interactions between inflammatory cytokines and tobacco smoke in the induction of endothelial dysfunction.
1. Introduction
Experimental evidence suggests that cigarette smoking damages vascular endothelium which plays a fundamental role in the genesis of pathological cardiovascular events. The mechanism(s), however, by which cigarette smoking alters endothelial homeostasis is still unclear. Cigarette smoking has been implicated in the activation of a complex inflammatory cascade resulting in the production of a variety of potent cytokines and chemokines, which in turn contribute to development of atherosclerotic plaques.
Many factors have been implicated in endothelial dysfunction, including the generation of reactive oxygen species (ROS), elevation of blood glucose, lipids and lipoproteins, and prostanoids produced via cyclooxygenase-2 (COX-2).1,2
The potential role of superoxide anion generated by NADPH oxidase is highly relevant in evaluating endothelial activation and dysfunction. NADPH oxidase consists of two membrane-bound components, Nox and p22phox, and several cytosolic regulatory subunits, including p40phox, p47phox, p67phox, and the small GTPase Rac. Upon enzyme activation, the cytoplasmic units translocate to the plasma membrane where they are co-assembled with Nox/p22phox. The resulting multisubunit complex transfers electrons from NADPH to molecular oxygen.3
Cyclooxygenases (COXs), mainly COX-1 and COX-2, convert arachidonic acid into the cyclic endoperoxide prostaglandin H2 (PGH2), which in turn is converted by specific enzymes into a variety of bioactive prostanoids, such as thromboxane A2, prostacyclin, and prostaglandin E2. Of relevance in this context is the observation that increased COXs, mainly COX-2, have been implicated in atherosclerosis and are strongly expressed in atherosclerotic plaques4 but not in normal blood vessels.
As yet, the interrelation(s) between cigarette smoking and circulating inflammatory cytokines, endothelial-derived ROS and COX-2 expression products have not been examined. We hypothesized that circulating component(s) of cigarette smoke may interact with inflammatory cytokines in the blood, inducing alterations in endothelial function, specifically increasing production of ROS and expression of COX-2. Our data indicate that serum from smokers induces ROS generation in vascular endothelial cells (ECs) through activation of NADPH oxidase, leading to consequent enhanced COX-2 expression and increased prostanoid production via the p38 mitogen-activated protein kinase (p38MAPK)/Akt pathway.
2. Methods
Detailed descriptions of standard methods, buffers and reagents can be found in the Supplementary material online.
2.1 Subjects
Nine healthy male non-smoker subjects (NS) and nine active smokers (AS) (mean ages, 31.25 ± 5.19 and 32 ± 5.43, respectively) were requested to abstain from food or caffeinated drinks overnight. The mean cigarette exposure in smokers was 13 ± 4 cigarettes per day. All subjects were free of other major cardiovascular risk factors, were not taking any medication, and provided informed consent for the study. One hour prior to blood collection each AS smoked one cigarette. Blood for in vitro and biochemical studies was collected from all NS and AS, in accordance with the principles outlined in the Declaration of Helsinki, incubated at 37°C for 1 h and centrifuged at 1000 g for 20 min at 4°C. Individual sera were used in initial experiments. Subsequently serum pools were prepared by pooling equal volumes of sera from all the AS together (pooled AS serum) and sera from all the NS together, respectively (pooled NS serum), and these were aliquoted and immediately stored at−80°C until use.
2.2 Animal treatments
FVB mice 10–12 weeks old received 0.2 mL intravenous tail injection of phosphate buffered saline (PBS) or of interleukin-1β (IL-1β) (1 ng/kg) + tumour necrosis factor alpha (TNF-α) (2.5 ng/kg) with or without TS (0.64 puff/kg) (five animals for each group). After 2 h of treatment mice were anaesthetized and perfused with saline and aortas and carotids were isolated. Aortas were processed for RT–PCR and carotids were embedded in optimal cutting temperature compound and stored at −80°C for ROS detection.
2.3 Cell culture and treatments
Studies were performed using a transformed human bone marrow microvascular endothelial cell line (TrHBMEC) described previously5 which maintains a normal microvascular endothelial phenotype, and human umbilical vein endothelial cells (HUVECs), isolated from freshly and anonymously acquired umbilical cords.6 Informed consent was provided in accordance with the Declaration of Helsinki. See Supplementary material online for further details of cell culture and treatments.
Apocynin, DPI (Sigma), LY294002, or SB203580 (Calbiochem) were added 2 h before cell stimulation, while Interleukin-1 receptor antagonist (IL-1ra) and anti-TNF-antibodies (TNF-ab) (R&D Systems, Inc.,) were added at the same time as the serum stimulus.
2.4 Intracellular ROS formation in cells and in carotid artery tissue
Cells were loaded with 10 μmol/L 2′,7′-dichlorofluorescein diacetate (DCFH-DA, Sigma) and the ROS production was measured by the intensity of DCF emission at 525 nm (excitation 502 nm), in a Perkin–Elmer LS50B fluorescence spectrometer. Results are expressed as the difference in DCF fluorescence (arbitrary units).7 Frozen, enzymatically intact, 10 μm thick cross-sections of carotid were incubated with dihydroethidium (DHE; 10 μmol/L, Sigma) and the images, obtained with a laser scanning confocal microscope (Carl ZEISS), were quantified using Image J software.8 See Supplementary material online.
2.5 Preparation of tobacco smoke extract
Tobacco smoke (TS) extract was prepared using 2R4F research cigarettes. Mainstream smoke generated in a Borgwaldt RM-1 smoking machine was bubbled through sterile PBS using the Federal Trade Commission standard protocol that mimics a standardized human smoking pattern (duration, 2 s/puff; frequency, 1 puff/min; volume, 35 mL/puff). The final concentration of TS in cell culture medium was expressed as puffs/L medium. See Supplementary material online.
2.6 Western blotting
For western blotting of total cellular extracts, cells were lysed in buffer A. For phosphorylated proteins, total cellular extracts were prepared by sonication of cells in lysis buffer B.9 For membrane and cytosol extracts, cells were suspended in relaxation buffer, sonicated on ice and centrifuged. The supernatant was then ultracentrifuged and membrane fractions were washed in relaxation buffer and dissolved in Laemmli sample buffer.10 Blotting was performed as described.7 See Supplementary material online. Membranes were incubated with antibody against COX-2, p47phox (both, 1:500, Santa Cruz), Akt, phospho-Akt (Ser 473), p38MAPK, phospho-p38MAPK (1:1 000, all from Cell Signaling), β-actin (1:10 000, Sigma) served as internal standard. The blots shown are representative of four independent experiments.
2.7 Reverse transcriptase–polymerase chain reaction (RT–PCR and QRT–PCR)
Total cellular RNA was isolated from cells or aorta tissue with TRIzol Reagent (Sigma) according to the manifacturer's instructions. Then, 1 μg of RNA was reverse transcribed using SuperScriptTM II RNase H (Invitrogen). Semi-quantitative polymerase chain reaction (RT–PCR) were performed using Taq DNA Polymerase (Invitrogen) and real-time quantitative PCR (QRT–PCR) with the fluorescent dye SYBER Green (Bio-Rad Laboratories) using specific primers. See Supplementary material online.
2.8 Measurement of cytokines
Human IL-1β and TNF-α levels in serum were measured using commercially available high-sensitivity ELISA assays according to the manufacturer's instructions (R&D Systems).
2.9 Transfection of siRNA
Transfection of cells with siRNAs was performed using Transfection Reagent and medium to result in a final p47-phox siRNA duplex or control siRNA (all from Santa Cruz) concentration of 50 nmol/L in according to the manufacturer's instructions. See Supplementary material online.
2.10 Data analysis
Data are shown as mean +SD and are based on at least four independent experiments. Statistical analysis was performed by Student's test or analysis of variance (ANOVA) and Fisher's LSD post test when appropriate. P < 0.05 was considered significant.
3. Results
3.1 Cytokine levels in active smoker sera and in non-smoker sera in relation to endothelial dysfunction in vitro
We first measure the serum levels of two inflammatory cytokines, IL-1β and of TNFα, that play a key role in the pathogenesis of inflammatory disorders such as atherosclerosis.
Levels of IL-1β and of TNFα were higher (1.7- and 2.4-fold, respectively) in individual AS sera compared with NS sera (Figure 1A and B). In particular, the mean serum levels were 11.48 ± 2.98 and 30.97 ± 7.83 ng/L in AS and 7.59 ± 3.12 and 14.36 ± 5.37 ng/L in NS subjects for IL-1β (P < 0.05) and TNFα (P < 0.01), respectively. A highly significant correlation between the serum levels of these two cytokines was observed (r = 0.818, P = 0.001) (see Supplementary material online, Figure S1A).
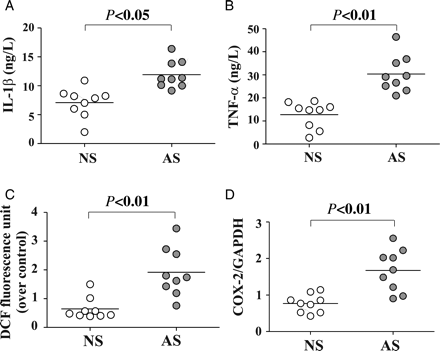
Serum cytokine levels in individual active smokers (AS) and non-smokers (NS) and effect of serum from individuals on ROS generation and COX-2 in endothelial cells. Serum concentration of (A) IL-1β, and (B) TNF-α in AS (filled circle) and NS (circle). Confluent TrHBMECs were incubated with 1% human serum from individual AS or NS, and (C) intracellular ROS production was detected after 5 min incubation, and (D) COX-2 mRNA was analysed by QRT–PCR after 1 h serum treatment.
We next examined the effect of sera on endothelial dysfunction in vitro in terms of ROS production and COX-2 expression.
To assess whether the content of ROS in serum of AS subjects was comparable with that of NS, in preliminary experiments the serum level of H2O2 were measured in each individual subject. We observed that the levels of H2O2 in the sera of AS were similar to those of NS (data not shown).
Furthermore, while mean fluorescence of TrHBMEC in the DCF assay, a reflection of intracellular ROS generation, rose only from 0.423 ± 0.111 A.U. in resting cells to 0.642 ± 0.376 in cells exposed for 5 min to 1% serum from each NS subject, it increased markedly to 1.917 ± 0.837 A.U. in cells exposed for 5 min to 1% serum from each AS subject (Figure 1C). Concomitant with ROS generation the mean COX-2 mRNA increased in cells exposed to individual AS sera compared with cells exposed to NS sera (1.677 ± 0.575 vs. 0.770 ± 0.249, P < 0.01, respectively) (Figure 1D). Increased ROS production by TrHBMECs treated for 1 h with sera of either AS or NS positively correlated with COX-2 mRNA (r = 0.822, P = 0.006) (see Supplementary material online, Figure S1B).
A significant positive correlation was also found between in vitro ROS production by TrHBMECs exposed to AS sera and the serum levels of IL-1β (r = 0.723, P = 0.002) (see Supplementary material online, Figure S1C) or TNF-α (r = 0.781, P = 0.0002) (see Supplementary material online, Figure S1D). Similarly, levels of COX-2 mRNA in cells exposed to serum of either AS or NS correlated positively with serum levels of the two inflammatory cytokines measured (IL-1β: r = 0.707, P = 0.002; TNF-α: r = 0.772, P = 0.0003) (see Supplementary material online, Figure S1E and F).
Our results showed that AS serum clearly stimulates ROS production and COX-2 expression in ECs.
3.2 Role of inflammatory cytokines IL-1β and TNF-α on ROS production and COX-2 expression by endothelial cells exposed to AS serum
When primary endothelial cells (HUVECs) and TrHBMEC were incubated with increasing amounts of pooled AS serum, ROS production and COX-2 expression both increased in a concentration-dependent manner, but not after exposure to pooled NS serum (Figure 2A–D and see Supplementary material online, Figure 2A–D).
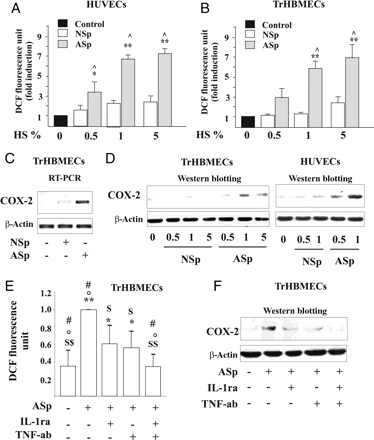
Effects of IL-1β and TNF-α present in smokers' serum on endothelial dysfunction in endothelial cells. Concentration-dependent effect of the pool of serum from nine AS (ASp) or pool of serum from nine NS (NSp) subjects on (A and B) ROS production. (C) COX-2 mRNA levels were detected by RT–PCR after 1 h exposure to ASp serum or NSp serum. (D) COX-2 protein expression was determined after 6 h of treatment with ASp serum or of the NSp serum. (E) Intracellular ROS generation and (F) COX-2 protein expression in TrHBMECs pre-incubated with IL-1ra and/or TNF-ab and then stimulated with ASp. *P < 0.05 and **P < 0.01 vs. control; ^P < 0.01 vs. NSp; $P < 0.05 and $ $P < 0.01 vs. ASp; °P < 0.05 vs. ASp + IL-1ra; #P < 0.05 vs. ASp + TNF-ab.
Increased ROS formation was detectable after 5 min' incubation with 1% pooled AS serum (Figure 2A and B) and augmented over 15 min (see Supplementary material online, Figure S2A). Moreover, 1% pooled AS serum up-regulated COX-2 mRNA (Figure 2C and see Supplementary material online, Figure S2B) and then induced expression of COX-2 protein (Figure 2D and see Supplementary material online, Figure S2C and D).
To assess whether inflammatory cytokines in AS caused increased ROS generation and COX-2 up-regulation, immune-neutralization experiments were carried out. Treatment of cells with IL-1ra (500 μg/L)11 or anti-TNF antibody (TNF-ab, 1 mg/L)12 reduced ROS formation (Figure 2E) and COX-2 protein (Figure 2F and see Supplementary material online, Figure 2E) in TrHBMEC exposed to pooled AS serum. In addition, treatment of TrHBMEC with IL-1ra and TNF-ab combined completely suppressed production of ROS (Figure 2E) and prevented induction of COX-2 expression by pooled AS serum (Figure 2F and see Supplementary material online, Figure S2E).
Collectively, these data indicate that IL-1β and TNF-α present in serum from AS are important modulators of EC activation.
3.3 TS requires low amounts of both IL-1β and TNF-α to induce endothelial dysfunction
We hypothesised that ‘pathologic’ concentrations of cytokines IL-1β and TNF-α in blood by themselves are not sufficient to induce endothelial ROS generation and COX-2 expression, two measures of endothelial dysfunction. Rather, cooperation between inflammatory cytokines and TS is needed for endothelial damage. To address this issue, HUVEC were incubated with a cocktail of both cytokines IL-1β (10 or 20 ng/L) and TNF-α (25 or 50 ng/L) alone or combined with low concentrations of TS (6.4 puffs/L) (Figure 3A and C and see Supplementary material online, Figure S3C). These concentrations of cytokines were chosen to include the range observed in the serum of smokers. We observed that neither TS alone nor either single cytokine altered ROS production by ECs (Figure 3B and see Supplementary material online, Figure S3A), whereas a small increase in ROS was detected when ECs were incubated with each of the dual combinations TS/IL-1β (20 ng/L), TS/TNF-α (50 ng/L), IL-1β (10 ng/L)/TNF-α (25 ng/L), or IL-1β (20 ng/L)/TNF-α (50 ng/L) (Figure 3A and B and see Supplementary material online, Figure S3A). In contrast, the simultaneous exposure of TrHBMECs to the triple combination of TS, IL-1β, and TNF-α (TS/IL-1β/TNF-α) resulted in a significant increase in ROS generation compared with any of the dual stimuli, TS/IL-1β, TS/TNF-α, or IL-1β/TNF-α (Figure 3B). Similar results were obtained using HUVEC (see Supplementary material online, Figure S3A). Doubling the concentration of IL-1β and TNF-α in combination with TS did not further modify ROS production (Figure 3A). High concentration of cytokines plus TS caused a seven-fold increase in ROS production above that of the control cells that was maximal at 5 min, progressively declining thereafter (four-fold increase vs. control at 15 min) although remaining significantly elevated (see Supplementary material online, Figure S3B).
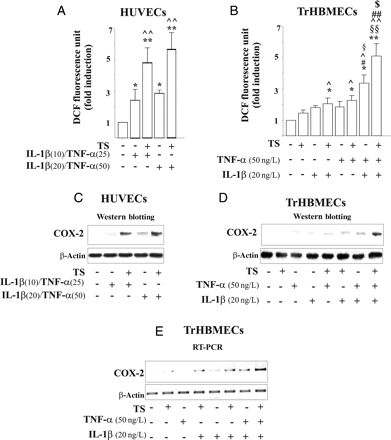
Effects of IL-1β and TNF-α combined with TS on endothelial dysfunction. (A and B) ROS generation after 5 min' stimulation (*P < 0.05 and **P < 0.01 vs. control, ^P < 0.05 and ^^P < 0.01 vs. TS, #P < 0.05 and #P < 0.01 vs. IL-1β, §P < 0.05 and §§ vs. TNF-α, and $P < 0.05 vs. IL-1β/TNF-α). (C and D) COX-2 protein and (E) COX-2 mRNA levels after stimulation with TS, TNF-α and/or IL-1β.
We next assessed the potential effects of the interaction of TS with inflammatory cytokines in inducing COX-2 in ECs. TS induced COX-2 expression when co-incubated with either low or high concentrations of cytokine cocktail (Figure 3C, see Supplementary material online, Figure S3C). Whereas a slight increase in both COX-2 protein and mRNA occurred in ECs exposed to the combinations of TS/IL-1β, TS/TNF-α, or IL-1β/TNF-α, addition of TS to IL-1β/TNF-α combined markedly augmented induction of COX-2 (Figure 3C–E, see Supplementary material online, Figure S3C–G).
We next evaluated the effect of treatment of a cocktail of cytokines (IL-1β and TNF-α) and TS extract in vivo in terms of ROS generation and COX-2 expression. Notably, tail vein injection of TS plus IL-1β/TNF-α increased both ROS generation and COX-2 mRNA levels in carotid and aorta tissue, respectively, compared with control animals or mice treated only with IL-1β/TNF-α (Figure 4A–C).
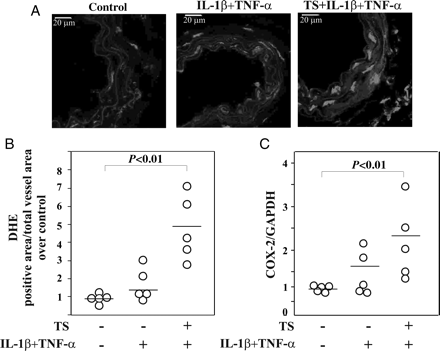
Effect of TS and/or IL-1β/TNF-α on ROS production and COX-2 expression in vivo. (A) Representative carotid cross-section labelled with the fluorescent dye DHE. Scale bars = 20 μm and (B) Quantitative fluorescent dye DHE of carotid after 2 h of tail vein injection of stimulus. (C) COX-2 mRNA of aortic tissue analysed by QRT–PCR after 2 h of treatment. (five animals per group).
These data indicate that cooperation between TS and inflammatory cytokines is necessary for induction of endothelial dysfunction manifested as increased ROS production and COX-2 expression.
3.4 Down-regulation of p47phox reduces ROS generation and COX-2 expression induced by pooled AS serum and TS/IL-1β/TNF-α
Evidence suggests that stable compounds present in cigarette smoke increase NADPH oxidase activity within cells to produce ROS.13–15 Therefore, we assessed the translocation of p47phox from cytoplasm to the plasma membrane as a marker of the in vitro activation of NADPH oxidase.
Under our conditions p47phox was detected mainly in cytoplasm both in resting TrHBMECs and in cells treated for 15 min with pooled NS serum, whereas p47phox mostly associated with the plasma membrane in cells exposed to pooled AS serum (Figure 5A, see Supplementary material online, Figure S4A).
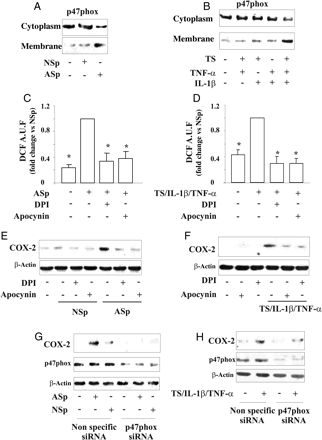
p47phox translocation and role of NADPH oxidase in the regulation of ROS generation and COX-2 expression induced by pooled serum and TS/IL-1β/TNF-α. Effect of (A) NSp serum or ASp serum, and (B) TS, IL-1β and/or TNF-α. TrHBMECs were pre-treated with (C–F) DPI or apocynin, or (G and H) transfected with non-specific siRNA or p47phox siRNA and then stimulated with pooled serum, or TS/IL-1β/TNF-α. (C and D) Intracellular ROS generation. (*P < 0.01 vs. ASp or TS/IL-1β/TNF-α). (E–H) COX-2 expression.
The simultaneous exposure of TrHBMECs to all three stimuli (TS/IL-1β/TNF-α) strongly decreased the cytoplasm amount of p47phox subunit and increased its association with the plasma membrane (Figure 5B, see Supplementary material online, Figure S4B).
Experiments were then performed to assess the contribution of NADPH oxidase to ROS production when TrHBMEC were exposed to pooled AS serum or TS/IL-1β/TNF-α. Cells incubated with 5 μmol/L DPI or 15 μmol/L apocynin, two inhibitors of NADPH oxidase, and treated with pooled AS serum or TS/IL-1β/TNF-α markedly decreased ROS generation (Figure 5C and D). Since ROS can modulate EC gene expression,16 we postulated that ROS produced through activation of NADPH oxidase regulate COX-2 expression. Indeed, inhibition of endothelial NADPH oxidase activity by DPI or apocynin prevented COX-2 induction either by pooled AS serum (Figure 5E, see Supplementary material online, Figure S4C) or by TS/IL-1β/TNF-α (Figure 5F, see Supplementary material online, Figure S4D) in TrHBMEC and in HUVEC (see Supplementary material online, Figure S4E–H).
Our inhibitor data obtained strongly suggest that a p47phox-dependent NADPH oxidase pathway is involved in COX-2 up-regulation. Furthermore, neither allopurinol (100 μmol/L) nor rotenone (1 μmol/L) had any effect on COX-2 expression (data not shown), indicating that neither xanthine oxidase nor the mitochondrial respiratory complex significantly modulate COX-2 in ECs under these conditions.
Finally, to confirm the relationship between NADPH oxidase-mediated ROS production and COX-2 expression, experiments were carried out using siRNA targeting NADPH oxidase subunit p47phox or a random sequence as control. Knockdown of p47phox by p47phox siRNA dramatically reduced ROS production (data not shown) and COX-2 expression in TrHBMECs exposed to the pooled AS serum (Figure 5G, see Supplementary material online, Figure S4I) or to TS/IL-1β/TNF-α (Figure 5H, see Supplementary material online, Figure S4L).
These data suggest that NADPH oxidase plays a key role in inducing endothelial dysfunction in smokers.
3.5 Akt and p38MAPK regulate endothelial COX-2 expression in smokers
Since we and others recently showed that induction of COX-2 in ECs by TS components involves activation of Akt9 and p38MAPK (p38),7 the role of phosphorylation of these kinases was also addressed here. Exposure of TrHBMEC to pooled AS serum or to TS/IL-1β/TNF-α for 10 min resulted in phosphorylation of both Akt at serine 473 and p38 (Figure 6A and B, see Supplementary material online, Figure S5A and B). In contrast, pooled NS serum or the cytokine combination IL-1β/TNF-α did not affect protein phosphorylation (Figure 6A and B, see Supplementary material online, Figure S5A and B).
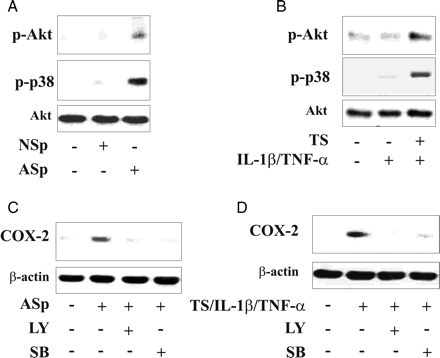
Role of Akt and p38MAPK on COX-2 expression in endothelial cells incubated with different stimuli. After exposure to pooled AS/NS serum, or TS and/or IL-1β/TNF-α, or TS/IL-1β/TNF-α, endothelial cells were lysed and processed for (A and B) phosphorylation of Akt (p-Akt) and p38MAPK (p-p38). In (C and D) TrHBMECs were preincubated in the presence or absence of LY294002 (LY), SB203580 (SB) for 2 h then stimulated with the ASp serum or TS/IL-1β/TNF-α and processed for COX-2 protein.
Pre-treatment of cells with LY294002 (LY: 2.5 μmol/L), a specific inhibitor of PI3K, or SB203580 (SB: 10 μmol/L), a specific inhibitor of p38, markedly reduced COX-2 expression induced by pooled AS serum or by TS/IL-1β/TNF-α in both TrHBMEC (Figure 6C and D, see Supplementary material online, Figure S5C and D) and HUVEC (see Supplementary material online, Figure S5E–H).
Thus, both Akt and the p38 pathway are involved in the regulation of COX-2 in ECs exposed to smoke components.
3.6 ROS induced by NADPH oxidase modulate the Akt/p38MAPK pathway
To identify a possible interaction among NADPH oxidase, Akt and p38, TrHBMECs were pre-treated with apocynin, DPI, LY, or SB and then exposed to pooled AS serum or TS/IL-1β/TNF-α. While both NADPH oxidase inhibitors (apocynin and DPI) and SB reduced the activation of Akt and p38, LY prevented only the Akt phosphorylation and had no effect on the p38 phosphorylation mediated by pooled AS serum or by TS/IL-1β/TNF-α (see Supplementary material online, Figure S6A–D).
Together, these data suggest that TS in association with inflammatory cytokines, induces phosphorylation of p38 and Akt via an NADPH oxidase dependent pathway.
4. Discussion
Smoking is a major risk factor for many human diseases, and is associated with generalized endothelial dysfunction.17
We have previously demonstrated that aqueous extracts of cigarette smoke interact with inflammatory cytokines to induce endothelial perturbation, in term of increased production of nitric oxide (NO), expression of intercellular cell adhesion molecules, monocyte adhesion, increased vascular permeability, ROS generation, and COX-2 expression.7,9 In this study, we show that inflammatory cytokines present in serum in concert with cigarette smoke induce endothelial dysfunction as indicated by increased ROS generation and COX-2 expression in vivo and in vitro. We also show that smokers' serum up-regulates ROS production in endothelium by activation of NADPH oxidase with consequent induction of COX-2 expression via the p38MAPK/Akt pathway.
Cytokines TNF-α and IL-1β both play critical roles in the pathogenesis of inflammatory disorders, and recent studies have linked certain polymorphisms in TNF-α or IL-1β with altered risks of coronary artery disease.18–20 In addition, several studies have associated smoking risks with changes in the expression of these cytokines21,22 emphasizing that short-term exposure to cigarette smoke in vivo is sufficient to increase IL-1β and/or TNF-α.9,23
In the present study, we observed that the levels of IL-1β and TNF-α in the serum of smoker subjects are elevated compared with non-smokers and that simultaneous inhibition of IL-1β and TNF-α-signalling pathways prevents smoking-induced endothelial dysfunction. We consider that these cytokines originate in the lung, most likely in epithelial cells and in leucocytes. Indeed, exposure to cigarette smoke increases levels of both IL-1β and TNF-α IL-1β in bronchial lavage fluid24 and in circulating mononuclear cells in smokers more than in non-smokers.25
In our model, the levels of IL-1β and TNF-α detected in smokers' serum failed to alter endothelial function alone or together, but when combined with saline-soluble smoke components promptly induced endothelial dysfunction. The critical role of cytokines in combination with yet undefined smoke components as observed in our work confirms and extends the results of studies, by ourselves and others, showing an important interaction between these cytokines and smoking in experimental animals or in humans.9,21,24,26,27
The role of IL-1β and TNF-α activation pathways in pathophysiological effects of smoking was previously described by Churg in IL-1 receptor and TNF-α receptor knockout mice.24 In particular, Churg et al.24 showed that blocking IL-1 or TNF-α receptors protects against acute smoke-mediated increases in inflammatory cells present in bronchial lavage fluid. The results in our study were consistent with this animal model: the effects of smokers' serum on human ECs were completely prevented by a combination IL-1ra and anti-TNF (TNF-ab).
Recently, Hart et al.21 showed that cigarette smoke condensate or benzo(a)pyrene induce the activation of the promoter of the IL1b gene presenting a specific polymorphism at -31T/C in human lung epithelial cells, and Hou et al.,22 showed that in smokers, the −806C > T polymorphism of TNF-α was associated with myocardial infarction, suggesting a significant interaction between IL-1β and TNF-α genes and smoking status.
The hypothesis previously proposed that inflammatory conditions can amplify and accelerate vascular dysfunction induced by smoke7,9 is confirmed by the data presented here. Indeed, cigarette smoke is known to regulate the expression and activity of COX-29,15 as well as the production of ROS15,28–30 in various cell types and animal model. We now provide the first direct evidence that cytokines present in the serum of smokers in association with TS components, induce ROS generation and increase COX-2 expression in human vascular ECs treated with serum from smokers.
Another pertinent observation is that serum from active smokers induces translocation of the p47phox subunit of NADPH oxidase from cytosol to plasma membrane. This confirms in a human system, prior published data using rat carotid arteries31 and smooth muscle cells.13,15 Moreover, using two pharmacological inhibitors (apocynin and DPI) and siRNA knockdown of p47phox, we show that inhibition of ROS generated via NADPH oxidase reduces the expression of COX-2 induced by serum from active smokers.
Consistent with our previous data obtained in cardiovascular tissue of mice exposed to cigarette smoke,7,9 we found that serum from active smokers not only increases COX-2 expression, but also induces the phosphorylation of p38 MAPK and Akt. This simple model allows us to investigate the signalling pathways involved in smoking-induced damage in cultured vascular cells, and to identify the signal transduction pathway that mediates the activation of NADPH oxidase, p38 MAPK, and Akt leading to enhanced COX-2 expression. It is also likely that Akt activation leads to the increase of NO generation as we previously showed for mouse endothelium. NO interacts with ROS to produce peroxynitrite (ONOO−), a powerful oxidant that also depletes vasoprotective NO. Indeed, previous studies showed not only that NO bioavailability is decreased in smokers,32,33 but also that elevated levels of nitroxidative biomarkers such as plasma levels of F-isoprostanes34 and protein 3-nitrotyrosine35 are detected in smokers. Remarkably oxidative alteration,29 inhibition of NO availability36 and COX-2 over-expression37 all can participate in the genesis of atherosclerosis.
In summary, our data show for the first time that serum from smokers modulates endothelial COX-2 expression by increasing NADPH oxidase-mediated ROS generation, with consequent activation of p38MAPK and Akt. Concomitantly, we provide evidence that the cytokines present in the serum of smokers interact with smoke components to up-regulate inflammatory gene expression in vascular ECs. This last key point emphasizes how the combination of smoking and inflammation is necessary to produce endothelial injury. The observation that serum of individual smoker subjects induces EC activation, may be of particular relevance for the development of a new assay aimed at defining the pro-atherogenic capacity of smoking in humans.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by Reti FIRB-2005-RBPR05NWWC
Acknowledgements
We are grateful to A. Ieraci for useful comments after reading this manuscript.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}