





Abstract
MicroRNAs (miRNAs) regulate various cardiac processes including cell proliferation and apoptosis. Pioglitazone (PIO), a peroxisome proliferator-activated receptor (PPAR)-γ agonist, protects against myocardial ischaemia–reperfusion (IR) injury. We assessed the effects of PPAR-γ activation on myocardial miRNA levels and the role of miRNAs in IR injury.
We evaluated the expression changes of miRNAs in the rat heart after PIO administration using miRNA arrays and then confirmed the result by northern blot. miR-29a and c levels decreased remarkably after 7-day treatment with PIO. In H9c2 cells, the effects of PIO and rosiglitazone on miR-29 expression levels were blocked by a selective PPAR-γ inhibitor GW9662. Downregulation of miR-29 by antisense inhibitor or by PIO protected H9c2 cells from simulated IR injury, indicated as increased cell survival and decreased caspase-3 activity. In contrast, overexpressing miR-29 promoted apoptosis and completely blocked the protective effect of PIO. Antagomirs against miR-29a or -29c significantly reduced myocardial infarct size and apoptosis in hearts subjected to IR injury. Western blot analyses demonstrated that Mcl-2, an anti-apoptotic Bcl-2 family member, was increased by miR-29 inhibition.
Downregulation of miR-29 protected hearts against IR injury. The modulation of miRNAs can be achieved by pharmacological intervention. These findings provide a rationale for the development of miRNA-based strategies for the attenuation of IR injury.
1. Introduction
MicroRNAs (miRNAs) are small non-coding molecules that negatively regulate gene expression via translational repression or mRNA degradation.1–3 Recent studies have shown that miRNAs control diverse aspects of heart disease, including hypertrophy, remodelling, heart failure, and arrhythmia,2–7 suggesting that there is a therapeutic potential for miRNA treatment of cardiovascular disease. The objective of the current study was to examine the role of miRNAs in the regulation of cell death and apoptosis in ischaemia–reperfusion (IR) injury. In particular, we proposed that specific miRNA can be modulated by pharmacological intervention. An alternative strategy is the antisense inhibitor techniques, which employs artificially designed oligonucleotides complementary specifically to the miRNAs. However, it remains a challenge to bring miRNAs antagonists from bench-to-bedside in clinical trials because of methods of delivery and the fact that specificity of targeting miRNA is difficult to achieve. Targeting miRNAs by pharmacological approach may represent a window of opportunity to apply miRNA therapy in the clinic setting. In this study, we focused on the interaction between miR-29s and pioglitazone (PIO), a peroxisome proliferator-activated receptor (PPAR)-γ agonist. PIO reduces myocardial infarct size (IS) in experimental animals.8–12 It was suggested that PIO limits IS by increasing the phosphorylation of the pro-survival kinase Akt.8,9 We found that PIO decreased myocardial levels of miR-29a and miR-29c. We then used artificially designed oligonucleotides, with either mimicking sequences or antisense sequences, to study the direct effects of these two miRNAs on IR injury and to determine whether the miR-29 mimicking oligonucleotides block the protective effects of PIO. As phosphoinositol-3-kinase (PI3K)10 and Mcl-1,11 an anti-apoptotic Bcl-2 family member, are putative targets of miR-29, we assessed the effects of inhibiting miR-29 on these pro-survival proteins.
2. Methods
2.1 Animal care
All animals received humane care in compliance with The Guide for the Care and Use of Laboratory Animals, published by National Institutes of Health (NIH Publication No. 85-23, Revised 1996). The study was approved by the University of Texas Medical Branch Institutional Animal Care and Use Committee.
2.2 miRNA microarray
Rats received 7-day pre-treatment with PIO 5 mg/kg/day or water (control). PIO was suspended in 1 mL of water and given once a day by oral gavage. There were three rats in each group. Total RNA including small RNAs were extracted from the left ventricle by TRIzol reagent. Ten micrograms of RNA samples were sent to L.C. Sciences for miRNA microarray.12 Samples were enriched for small RNA. Each pair of samples (control and PIO) were labelled with Cys3 and Cys5 fluorescent dyes and hybridized to a single Atactic μParaFlo microfluidics chip that held all the 334 mature rodent miRNA probes. Perfectly matched and mismatched probes were used for quality control. The raw data represented the average of nine signal values for each miRNA on the array. The background was subtracted and normalized using LOWESS (locally weighted regression) method. A Student t-test was used to compare between control and PIO groups and t-values were calculated for each miRNA.
2.3 Northern blotting analysis
Rats received 3- or 7-day pre-treatments with the same group as discussed earlier. Total RNA was isolated using the TRIzol protocol (Invitrogene), and northern blot analyses were performed according to the manufacturer's instructions. Membranes were exposed using a chemiluminescence imaging system (Ultralum, Inc., Claremont, CA, USA). Expression levels were evaluated using the Typhoon Phosphor Imager software analysis (Amersham Biosciences, Piscataway, NJ, USA).
2.4 Determination of miR-29 expression level
The cardiac muscle H9c2 cells were maintained in complete growth medium, supplemented with 10% FBS and penicillin-G (100 IU/mL). Cells were maintained in tissue culture incubators at 37°C under 5% CO2 atmosphere. Cells were seeded at 1 × 106 per 100 mm plate and incubated until 60–70% confluent. Cells were treated with (i) vehicle (2% ethanol); (ii) 10 μM PIO; (iii) 1 μM GW9662 (a PPAR-γ inhibitor); (iv) 10 μM PIO + 1 μM GW9662; (v) 2 μM rosiglitazone (ROSI); or (vi) 2 μM ROSI + 1 μM GW9662. PIO and ROSI were dissolved in ethanol (final concentration 2%). Twenty-four hours later, cells were harvested and total RNA was isolated using the mirVana miRNA Isolation Kit (Ambion). miR-29 levels were measured using the mirVana qRT–PCR miRNA Detection Kit. Reactions containing mirVana qRT–PCR Primer sets were specific for rat miR-29. Data were normalized by evaluating U6 expression.
2.5 Transfection and treatment groups
Exponentially growing H9c2 cells were seeded at 1 × 106 per 100 mm plate or at 1 × 105 per 96-well plate overnight until 50% confluent. Then, cells were exposed to various agents for transfection experiments. In tube 1, 10 nM miR-29 inhibitor, mimic, and non-targeting controls were placed, and the transfection reagent placed in tube 2 with serum-free medium. The contents of tubes 1 and 2 were combined and incubated for 20 min at room temperature. Complete medium, together with the mixture from the above were then added to the plates after removing the old medium. Cells were incubated at 37°C with 5% CO2 for 24 h with antibiotic-free medium.
Protocol 1: (i) negative control for mimic (NC-mim); (ii) negative control for inhibitor (NC-inh); (iii) miR-29a-mimic (mim-29a); (iv) miR-29a-inhibitor (inh-29a); (v) miR-29c-mimic (mim-29c); (vi) miR-29c-inhibitor (inh-29c).
Protocol 2: (i) vehicle (2% ethanol); (ii) PIO (10 μM); (iii) PIO + mim-29a; (iv) PIO + mim-29c; (v) PIO +mim-29a + mim-29c.
2.6 Simulated IR
Twenty-four hours after transfection, cells were subjected to SIR: 16 h hypoxia and 2 h reoxygenation or 18 h incubation in normoxemic condition (NSIR). Hypoxia consisted of layering mineral oil over a thin film of hypoxic medium (pre-bubbled with N2 gas) that covered the cells for 16 h. The hypoxic medium was an HEPES-buffered medium that contained (in mmol/L) 139 NaCl, 4.7 KCl, 0.5 MgCl2, 0.9 CaCl2, 5 HEPES, pH 7.4. The hypoxia medium was replaced by fresh medium (complete growth medium) and followed by 2 h reoxygenation. SIR-induced cell death was measured by counting trypan blue-stained cells and expressed as a percentage of the total cells counted, based on the ability of live cells to exclude trypan blue. Cell viability was measured by 3-[4,5-yl]-2,5-diphenyltetrazolium bromide (MTT) cell respiration assay kit according to the manufacturer's instruction. The apoptotic nuclei were labelled using terminal deoxy-nucleotidyl transferase-mediated dUTP nick end-labelling (TUNEL) assay kit from Roche. The enzymatic activity of caspase-3 induced by various conditions was measured with the Caspase-3 Colorimetric Protease Assay Kit according to the manufacturer's protocol. Additional details are provided in the Methods section of the Supplementary material online.
2.7 MTT cell viability assay
Cell viability was measured by MTT cell viability assay. MTT cell respiration assay measures the mitochondrial capability to reduce MTT substrate in live cells. Cells were treated with 3-[4,5-yl]-2,5-diphenyltetrazolium bromide (MTT, 0.5 mg/mL) for 4 h at 37°C. The attached cells were lysed in 2-isopropanol containing 0.04 M HCl, and the amount of metabolized MTT was determined using a micro-plate reader. Data are expressed as a percentage of the control NSIR group.
2.8 TUNEL assays
To determine myocardial apoptosis quantitatively, mice were studied in an additional experiment using TUNEL staining. Mice received the same treatment as discussed earlier and subjected to 30 min ischaemia and 4 h reperfusion. At 4 h, the coronary artery was re-ligated, and blue dye was injected to the right ventricle to identify the myocardial area at risk. Hearts were removed and sliced and stained with 2,3,5-triphenyl-tetrazolium-chloride (TTC) to differentiate infarcted (TTC unstained) from viable myocardium (TTC stained). The border zone was identified as Evans blue unstained and TTC stained. A 2 mm section of myocardium tissue from the border zone was fixed in 4% formalin solution and embedded in paraffin. Then, 5 μm thickness sections cut from each tissue block were mounted on superfrost slides and dried overnight at 37°C. Immunohistochemical procedures for detecting apoptotic cardiomyocytes were performed using Cardio-TACS kit (Trevigen; Gaithersburg, MD, USA) according to the manufacturer's instructions as described previously.13 In this procedure, apoptotic nuclei are stained blue. Nuclear Fast Red was used as a counterstain, which provided super contrast to the blue label in apoptotic cells. TUNEL-positive myocytes were determined by randomly counting 10 fields. The index of apoptosis was then determined (i.e. number of apoptotic myocytes/total number of myocytes counted × 100).
2.9 Statistical analysis
Data are presented as means ± standard errors of means. One-way analysis of variance (ANOVA) with Sidak correction for multiple comparisons was applied to compare the different groups. P < 0.05 was considered statistically significant.
3. Results
3.1 miR-29 expressions are downregulated by PIO
We first identified the miR-29 family as a candidate miRNA that is downregulated by PIO using miRNA array screening. As shown in Figure 1A and B, PIO downregulated myocardial levels of miR-29a, -29b, -29c and upregulated miR-15. On the basis of their basal expression level in the control hearts and the changes induced by PIO, we decided to focus on miR-29a and -29c. Northern blot analyses of RNA from rat left ventricle confirmed decreased expression of miR-29a and miR-29c after PIO administration (Figure 1C–F).
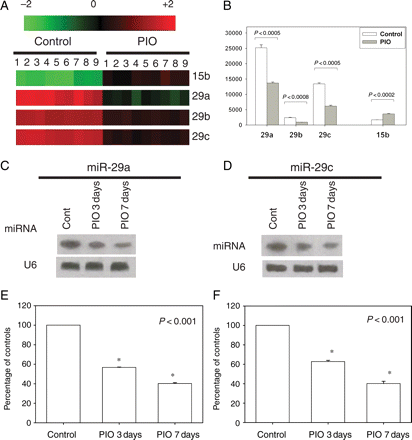
MiRNA expression changes after administration of PIO (5 mg/kg/days) or water for 7 days (there were three rats in each group). (A) Log2 values of each PIO/sham pair of miRNA microarray signal were displayed in a heat-map generated by TIGR Multiexperiment Viewer software. Red indicates upregulation; green, downregulation; black, no change. The bar code on the top represents the colour scale of the log2 values. (B) Bar graph indicating the fold change in expression in PIO group compared with the baseline of the miRNAs of interest. (C and D) Northern blot showed the time course effect of PIO on miR-29a and -29c expression levels in rat hearts. (E and F) The densitometry analysis of northern blot. The data represented the average of nine signal values for each miRNA on the array. *P < 0.05 vs. control.
Both CD36 and AP2 are PPAR-γ-dependent genes encoding proteins involved in lipid transport and storage.14,15 We used AP2 and CD36 as markers of PPAR-γ activation. Using RT–PCR, we found that both AP2 and CD36 mRNA levels were increased by PIO after 7-day administration (Supplementary material online, Figure S1).
3.2 The effect of PIO and ROSI on miR-29 is blocked by GW9662
We assessed the effect of GW9662, a selective PPAR-γ inhibitor, on the downregulation of miR-29 by two different PPAR-γ activator thiazolidinediones (PIO and ROSI) in H9c2 cells by qRT–PCR. As shown in Supplementary material online, Figure S2A and B, qRT–PCR revealed that miR-29a and miR-29c were highly expressed in H9c2 cells and downregulated by both PIO and ROSI. The inhibitory effect of PIO and ROSI on miR-29 expression was blocked by GW9662, a PPAR-γ inhibitor. According to real-time PCR analyses, both AP2 and CD36 mRNA levels were increased by PIO and ROSI and blocked by GW9662 in H9c2 cells (Supplementary material online, Figure S2C and D), suggesting that the effect of PIO or ROSI on miR-29 was mediated by PPAR-γ activation.
3.3 Downregulation of miR-29 promotes cell viability and prevents cell death induced by SIR
To assess the functional consequences of silencing or overexpression of miR-29 in SIR, antisense inhibitor and mimic were transfected into H9c2 cells. Northern blot assays confirmed that the expression levels of miR-29a and c were increased three-fold by mim-29a and mim-29c compared with negative control. In contrast, miR-29a and c levels were decreased by the specific inh-29a and inh-29c (Figure 2A and B). After transfection, cells were subjected to SIR or NSIR. Cell viability, assessed by MTT assay, was reduced by SIR injury (Figure 2C and D).
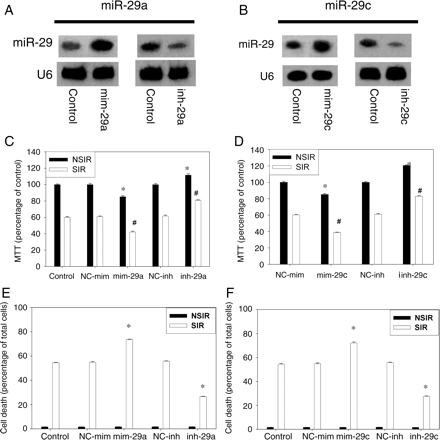
Effect of miR-29 on the viability of H9c2 cells subjected to SIR or NSIR. (A and B) miR-29 expression levels were assessed by northern blot after 24 h transfection of mimic (mim-29) or inhibitor (inh-29). There were four samples in each group. (C and D) MTT viability in cells subjected to SIR or NSIR. Results are expressed as percentage of NC-mim-NSIR, taken as 100% (eight independent experiments were performed). Overall, there were significant differences among the treatment groups in both the cells not exposed to SIR (NSIR, P < 0.001) and those exposed to SIR (P < 0.001). *P < 0.05 vs. NC-mim-NSIR. #P < 0.05 vs. NC-mim-SIR. (E and F) Cell death (six independent experiments were performed for each group). There was no difference among the treatment groups in cells not exposed to SIR (NSIR; P = 0.974 for miR-29a and P = 0.828 for miR-29c). Among cells subjected to SIR, there were significant differences among groups (P < 0.001 for miR-29a and P < 0.001 for miR-29c). *P < 0.05 vs. control SIR.
Overexpression of miR-29a or -29c by mimics reduced cell viability, whereas inhibition of miR-29a or -29c by inh-29a or inh-29c increased cell viability in both SIR and NSIR condition (Figure 2C and D). Negative controls (NC-mim and NC-inh) had no effect.
Total cell death was assessed by trypan blue staining. mim-29a and mim-29c augmented cell death induced by SIR. In contrast, inh-29a and inh-29c significantly reduced cell death in cells exposed to SIR (Figure 2E and F).
3.4 Overexpression of miR-29a and miR-29c blocks the protective effect of PIO
Because in the in vitro model we had to dissolve PIO and ROSI in 2% alcohol, we tested whether 2% alcohol altered cell viability. As depicted in Supplementary material online, Figure S3, 2% alcohol had no effect on cell viability. PIO enhanced the viability of cells subjected to SIR (Figure 3A). The protective effect of PIO was partially blocked by transfection of mim-29a or mim-29c alone. This effect was completely abolished by co-transfection with both oligonucleotides.
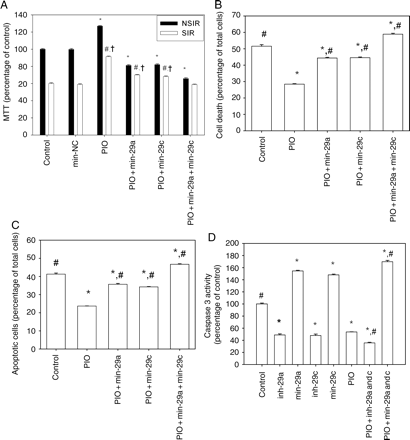
MiR-29 is involved in the protective effect of PIO against SIR. H9c2 cells were treated with: (i) control (2% ethanol); (ii) negative control mim (mim-NC); (iii) PIO 10 μM; (iv) PIO + mim-29a; (v) PIO + mim-29c, and (vi) PIO + mim-29a + mim-29c. (A) Cell viability was assessed by MTT assay. Results are expressed as percentage of control, taken as 100% (eight independent experiments for each group). Overall, there were significant differences among groups, both in cells not exposed to SIR (NSIR, P < 0.001) and in cells exposed to SIR (P < 0.001). *P < 0.05 vs. control-NSIR. #P < 0.05 vs. control-SIR. †P < 0.05 vs. PIO + mim-29a + mim-29c-SIR. (B) The percentage of dead cells after exposure to SIR, assessed by trypan blue staining. Results represent means ± SEM of the per cent of positively stained cells (eight independent experiments for each group). Overall, there were significant differences among groups (P < 0.001). *P < 0.5 vs. control. #P < 0.05 vs. PIO. (C) Quantitative assay of apoptotic cells after exposure to SIR. Results are expressed as percentage of TUNEL-positive cells (six independent experiments for each group). Overall, there were significant differences among groups (P < 0.001). *P < 0.05 vs. control. #P < 0.05 vs. PIO. (D) Caspases-3 activity (four independent experiments for each group). Overall, there were significant differences among groups (P < 0.001). *P < 0.05 vs. control. #P < 0.05 vs. PIO.
Cell death was assessed by trypan blue staining. PIO significantly reduced death of cells exposed to SIR (Figure 3B). The protective effect of PIO was partially blocked by mim-29a and mim-29c and completely blocked by co-transfection with both mim-29a and mim-29c.
Apoptotic cell death was verified by TUNEL assay for DNA cleavage (Figure 3C). PIO significantly decreased the number of positively stained cells after exposure to SIR. Transfection with mim-29a or mim-29c, respectively, partially blocked the effect of PIO. Co-transfection with mim-29a and mim-29c completely blocked the anti-apoptotic effect of PIO. Representative samples of TUNEL staining are shown in Supplementary material online, Figure S5.
We measured caspase-3 activity in all groups subjected to SIR. Compared with the control group, inhibition of miR-29 by inh-29a or inh-29c caused a significant decrease in caspase-3 activity (Figure 3D). PIO also significantly reduced caspase-3 activity. The effect of PIO on caspase-3 activity was blocked by co-transfection of mim-29a and miR-29c, whereas it was augmented by co-transfection of both.
3.5 The effect of miR-29 on Mcl-1 in vitro
Among the more than 300 computationally predicted targets of miR-29, we identified Mcl-1 as a highly potential miR-29 target. The predictive binding sites on 3′-UTRs of mice or rats PTEN interact specifically with miR-29a and -29c, as shown in Supplementary material online, Figure S7.
Transfection of H9c2 cells with mim-29a or mim-29c produced a significant decrease in Mcl-1 protein levels (Figure 4A and C). Transfection with either inh-29a or inh-29c did not cause significant changes in Mcl-1 protein levels (Figure 4B and D). Co-transfection with inh-29a and -29C, however, increased the level by 67% (P < 0.001).
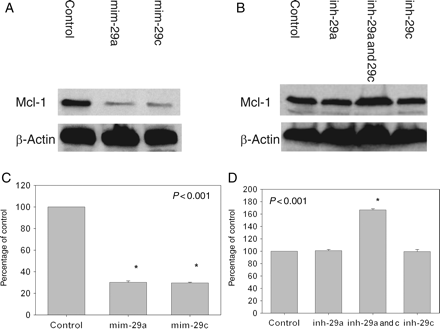
Effect of miR-29a and miR-29c on Mcl-1 levels in vitro. H9c2 cells were transfected with miR-29 mimicking or inhibitory oligonucleotides for 24 h. (A and B) Samples of immunoblots. (C and D) Densitometric analyses of Mcl-1 expression. *P < 0.05 vs. control. Results were representative of four independent experiments.
3.6 The effect of antagomir-29a and antagomir-29c on IS
Sixteen hours after the third injection, qRT–PCR showed effective suppression of miR-29a (Figure 5A) and miR-29c (Figure 5B) by antagomir-29a and 29c (anta-29a and anta-29c), respectively, but not by missense antagomir (misanta-29). A total of 24 mice were included (six per group). None of the mice died or were excluded. Body weight and the size of the area at risk were comparable among groups (Supplementary material online, Table S1). IS was significantly smaller in the anta-29a- and anta-29c-treated group than in the control group (receiving PBS injection), whereas misanta-29 had no effect (Figure 5C). Samples of IS are depicted in Supplementary material online, Figure S4. Thus, downregulation of miR-29a and c confers cardioprotection against IR injury in vivo.
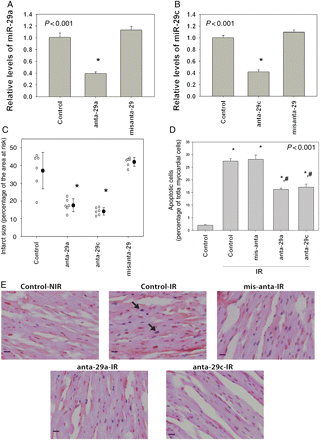
(A) Relative expression of miR-29a 16 h after the third injection of anta-29a or missense antagomir (misanta-29). (B) Relative expression of miR-29c 16 h after the third intravenous injection of anta-29c or missense antagomir (misanta-29). (C) Effect of anta-29a, anta-29c, and misanta-29 on myocardial IS (percentage of the ischaemic area at risk). There were significant differences among groups (P < 0.001). *P < 0.05 vs. the control group. (D) Paraffin-embedded sections of antagomir- or missense antagomir-pre-treated hearts subjected to 30 min ischaemia and 4 h reperfusion were stained by TUNEL to detect apoptosis. Immunolabelled nuclei of myocytes were determined by random counting of 10 fields per section. Each bar represents the mean ± SE of six hearts. Per cent of TUNEL-positive-stained cardiomyocytes in the ischaemic area at risk of mice hearts (control: sham-treated hearts not exposed to IR; control-IR: sham-treated hearts exposed to IR). There were six animals in each group. (E) Representative staining pattern. Arrows indicate TUNEL-positive nuclei which are stained blue. Magnification: ×20.
3.7 Detection of apoptosis by TUNEL staining
To examine whether anta-29a and anta-29c have anti-apoptotic action, TUNEL staining was performed in vivo. Heart tissue from sham-operated mice (Control-NIR) exhibited low levels of TUNEL-positive cells (2.0 ± 0.2%). After 30 min ischaemia and 4 h reperfusion, the percentage of TUNEL-positive cells in the border zone of the previously ischaemic area increased to 27.4 ± 1.0. Anta-29a (16.2 ± 0.5%) and anta-29c (17.1 ± 1.2%) attenuated this increase, whereas misanta-29 (28.2 ± 1.7%) had no effect (n = 6 per group) (Figure 5D). Representative myocardial samples are shown in Figure 5E. In our study, the border zones of post-ischaemic hearts that received anta-29a and anta-29c had fewer TUNEL-positive cardiomyocytes than control (IR), suggesting that the cardioprotective effect of antagomirs of miR-29 involves inhibition of cardiac apoptosis.
3.8 The effect of antagomir-29a and antagomir-29c on P-Akt, BAX, and Mcl-1 levels in vivo
In sham-operated mice, anta-29a and anta-29c induced mild increase in Mcl-1 levels (Supplementary material online, Figure S6A and B), but not in Ser-473 P-Akt or total Akt levels (Supplementary material online, Figure S6A, C and D). There were four mice in each group. Heart tissue from sham-operated mice (serve as control) or the border zone of the ischaemic area of mice undergoing IR protocol as discussed earlier was further investigated. There were four mice in each group. IR injury did not affect total Akt levels (Figure 6A and C). Anta-29a and 29c also had no effect on total Akt levels. Ser-473 P-Akt levels increased in the border zone after IR in the vehicle-treated mice (Figure 6A and D). Anta-29a and 29c augmented this increase. IR decreased myocardial levels of Mcl-1 in the border zone. On the other hand, anta-29a and anta-29c increased Mcl-1 levels after IR (Figure 6B and E). IR increased BAX level in the border zone, and Anta-29a and anta-29c attenuated this increase (Figure 6B and F).
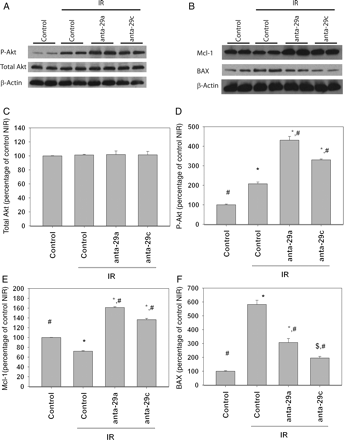
Effects of anta-29a and anta-29c on myocardial levels of total Akt, P-Akt, Mcl-1, and BAX. (A and B) Western blot analysis of P-Akt and total Akt from hearts treated with anta-29a and -29c and exposed to 30 min ischaemia and 4 h reperfusion. Aliquots of 100 mg of heart homogenates from mice heart were separated on SDS–PAGE and transferred to nitrocellulose membranes. (C and D) Densitometric analysis of total Akt, P-Akt, Mcl-1, and BAX levels (four independent experiments performed) *P < 0.001 vs. control-NIR; #P < 0.001 vs. control-IR; $P = 0.054 vs. control-NIR.
4. Discussion
The novel findings of this study are summarized as follows. First, we showed that miR-29a and miR-29c levels were downregulated by the PPAR-γ agonists PIO and ROSI. This effect was blocked by the PPAR-γ inhibitor GW9662. Second, we showed that miR-29a and miR-29c are involved in IR injury. Inhibition of miR-29a or miR-29c by antisense inhibitors reduced cell death induced by SIR injury. The protective effect of PIO against SIR injury was blocked by overexpressing miR-29. Finally, we used an in vivo model to show that intravenous injection of antagomir-29a or 29c limits myocardial IS and caused a significant decrease in apoptosis. For the first time, our results show an anti-apoptotic effect of miR-29 inhibition in IR injury, which can be regulated by PPAR-γ agonists.
Pharmacological agents inhibiting or blocking pro-apoptotic pathways during IR can exert a cardioprotective effect. Our work and that of others have shown that thiazolidinediones, such as PIO, limit myocardial IS.9,16–19 However, the underlying mechanisms of this protective effect are not fully understood. It was suggested that PIO limits IS by increasing the phosphorylation of the pro-survival kinase Akt.8,9 However, there is disagreement whether the ATP-sensitive K+ (KATP) channels and endothelial nitric oxide synthase are involved.8,16,20 In the present study, we performed an miRNA array to show an altered expression profile of miRNAs induced by PIO. We found that the miR-29 family was significantly downregulated in the rat heart after pre-treatment with PIO (Figure 1). The result was confirmed by northern blot. Therefore, miR-29a and 29c were further studied using an in vitro SIR model to determine the interaction between PIO and miR-29. RT–PCR showed that both PIO and another PPAR-γ activator ROSI decreased miR-29a and c levels (Supplementary material online, Figure S2). This effect was blocked by a PPAR-γ inhibitor, suggesting that this effect is dependent of PPAR-γ activation. PIO increased H9c2 cell viability and protected cells from SIR injury (Figure 3). Moreover, PIO attenuated the activation of caspase-3 following SIR. All these effects were blocked by overexpression of miR-29, suggesting an essential role for the involvement of miR-29 in the cardioprotective effect of PIO against IR injury. Our data have shown that alteration of miR-29 expression is a significant outcome after the administration of PIO, and modulation of miRNA can be achieved pharmacologically in a potentially clinically relevant fashion.
Increasing evidence suggests that miRNAs play pivotal roles in various heart diseases by altering transcription of important genes.2,7,21 Consistent with these reports, our data showed that downregulation of miR-29 by PIO, or by antisense inhibitors, helps H9c2 myocytes evade cell death induced by SIR injury. Transfection with miR-29a and -29c mimics decreased cell viability and induced apoptosis, whereas transfection with miR-29a and -29c antisense inhibitors reduced cell death and conferred resistance to IR injury (Figure 2). To confirm the in vitro results, we utilized an in vivo IR injury model and demonstrated that downregulation of miR-29 by antagomir limited myocardial IS and attenuated myocardium apoptosis in the risk area. It has been reported, however, that the extent of necrosis peaks at 24 h of reperfusion, and the number of apoptotic cells in the perinecrotic area progressively increases up to 72 h.22 Measurement of apoptosis at a single time point may be insufficient to determine the apoptosis during IR injury. Experiments to determine the effects of anti-miR-29 antagomirs on apoptosis at different time points after reperfusion and on left ventricular remodelling after infarction are needed.
miRNAs enforce their function through post-transcriptional silencing of RNA interference pathway. Mcl-1 has strong anti-apoptosis properties. Through bioinformatics algorithm analysis, we identified predictive miR-29-binding sites at 3′-UTR of Mcl-1 (Supplementary material online, Figure S7). Transfection of non-malignant cells with miR-29b antisense oligonucleotides increases Mcl-1 levels.11 Mcl-1 is involved in late ischaemic preconditioning of the heart.23 In this study, we found that a combination of antisense oligonucleotides for both miR-29a and miR-29c increased the levels of Mcl-1, whereas miR-29a or miR-29c mimic oligonucleotides alone reduced Mcl-1 levels. Anta-29a and anta-29c increased Mcl-1 levels in hearts exposed to IR injury, suggesting that the protective effect of miR-29 is associated with the modulation of expression of Mcl-1.
As Akt is involved in mediating ischaemic and pharmacological preconditioning, including myocardial protection by PIO,9,24 modulation of Akt phosphorylation by miR-29 could be part of the signalling pathway of myocardial protection by PIO. Indeed, our in vivo experiments showed that anta-29a and anta-29c increased myocardial levels of P-Akt (Figure 6). It has also been shown that miR-29 suppresses the p85γ regulatory subunit of PI3K.10 van Rooij suggested that miR-29s are associated with fibrosis-related mRNAs.25 Downregulation of miR-29 induced collagen expression both in vitro and in vivo.25 Long-term activation of pro-survival pathways, such as PI3K and Akt, leads to fibrosis and hypertrophy, whereas short-term activation may confer protection against IR injury.26–28
Other miRNAs have been reported to be involved in IR injury. For example, MiR-320 inhibition reduces myocardial IS in mice, whereas overexpression of miR-320 enhanced cardiomyocytes death and apoptosis. The putative target of miR-320 is heat-shock-protein-20.29 Downregulation of miR-199a de-represses hypoxia-inducible-factor-1α and Sitrulin-1 and induces preconditioning.30 It was reported that ischaemic preconditioning upregulates miRNA-1, miRNA-21, and miRNA-24. Injection of these miRNAs reduces IS and induces eNOS, heat-shock-transcription-factor-1, and heat-shock-protein-70.31 MiR-92a improved recovery of LV function in a mouse model of myocardial infarction.32
Taken together, our finding reveals an important role for specific miRNAs in the control of IR injury-induced cell death and points to miRNAs as potential drug targets for the treatment of heart disease. Inhibiting miRNAs by antisense strategies and especially by pharmacological approaches are likely to emerge as alternative and safe methods for conferring short- and intermediate-term protection against IR injury.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
The study was partially supported by Takeda Pharmaceuticals North America.
References
Author notes
These authors contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}