





Abstract
Thrombin activates protease-activated receptor 1 by proteolytic cleavage of the N-terminus. Although much research has focused on the activated receptor, little is known about the 41-amino acid N-terminal peptide (parstatin). We hypothesized that parstatin would protect the heart against ischaemia–reperfusion injury.
We assessed the protective role of parstatin in an in vivo and in vitro rat model of myocardial ischaemia–reperfusion injury. Parstatin treatment before, during, and after ischaemia decreased infarct size by 26%, 23%, and 18%, respectively, in an in vivo model of ischaemia–reperfusion injury. Parstatin treatment immediately before ischaemia decreased infarct size by 65% and increased recovery in ventricular function by 23% in an in vitro model. We then assessed whether parstatin induced cardioprotection by activation of a Gi-protein-dependent pathway. Gi-protein inactivation by pertussis toxin completely abolished the cardioprotective effects. The cardioprotective effects were also abolished by inhibition of nitric oxide synthase (NOS), extracellular signal-regulated kinases 1/2 (ERK1/2), p38 mitogen-activated protein kinase (p38 MAPK), and KATP channels in vitro. Furthermore, parstatin increased coronary flow and decreased perfusion pressure in the isolated heart. The vasodilatory properties of parstatin were confirmed in rat coronary arterioles.
A single treatment of parstatin administered prior to ischaemia confers immediate cardioprotection by recruiting the Gi-protein activation pathway including p38 MAPK, ERK1/2, NOS, and KATP channels. Parstatin exerts effects on both the cardiomyocytes and the coronary circulation to induce cardioprotection. This suggests a potential therapeutic role of parstatin in the treatment of cardiac injury resulting from ischaemia and reperfusion.
1. Introduction
Although reperfusion therapy for acute myocardial infarction reduces infarct size, improves left ventricular (LV) function, and reduces mortality, the full potential benefit is limited by acceleration of damage resulting from reperfusion or ischaemia–reperfusion injury. Several exogenous agents have been shown to limit myocardial ischaemia–reperfusion injury but have not made it into clinical practice.
With regard to ischaemia–reperfusion injury, both the cardiomyocytes and the coronary circulation may be affected. The injury to myocytes may directly reduce cardiac contractility. The injury to the coronary circulation may change coronary vascular tone and resistance and, therefore, affect coronary flow during the reperfusion period. Reduction of coronary flow may damage myocardial perfusion and this, in addition to ischaemia–reperfusion injury to the myocardium, may further damage myocardial function. This functional change may cause mortality and morbidity in cardiac surgery. Based on these considerations, in recent years, some studies have focused on ischaemia–reperfusion injury to the coronary circulation in addition to studies on injury to myocytes.1 These studies have been conducted by either measuring the coronary flow in the perfused heart2–4 or measuring endothelium-dependent relaxation in isolated coronary vessels.5,6
Thrombin cleaves the protease-activated receptor 1 (PAR1) to release a 41-amino acid peptide and activates the remaining G-protein-coupled receptor. Inhibition of PAR1 has been shown to be protective against myocardial ischaemia–reperfusion injury.7 However, activation of PAR1 has been shown to be important in vasculogenesis along with vasodilation.8,9 Although much research has focused on the activated receptor, little is known about the N-terminal 41-amino acid peptide that is cleaved off by thrombin. Recently, it has been demonstrated that the 41-amino acid peptide (parstatin) has endothelial effects;10 however, its role in myocardial ischaemia–reperfusion injury is unknown. Therefore, we hypothesized that parstatin would be cardioprotective during ischaemia–reperfusion injury by its effects on both the cardiomyocytes and the coronary circulation.
2. Methods
Male Sprague–Dawley rats at 8 weeks of age (250–300 g) used in this study received humane care in compliance with the ‘Guide for the Care and Use of Laboratory Animals’ published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). This project was granted approval by the local IACUC review board. The 41-amino acid peptide (M1GPRRLLLVAACFSLCGPLLSARTRARRPESKATNATLDPR41) named parstatin was synthesized by EZBiolab Inc. (Westfield, IN, USA). Synthesized peptides were purified by HPLC technology and to a purity of >95%.
2.1 Parstatin and cardioprotection studies in vivo
An in vivo anaesthetized rat model was used for these experiments with the general surgical protocol and determination of infarct size described previously.11 For infarct size studies, rats (n = 6/group) underwent 30 min regional ischaemia followed by 120 min reperfusion. Parstatin (1–25 µg/kg, iv) was injected 15 min prior to ischaemia. After the optimal dose was determined, a temporal response curve was determined by administering parstatin (10 µg/kg, iv) at 15 min after the onset of ischaemia, or 10 s after the onset of reperfusion in a separate series of experiments (n = 6/group).
2.2 Parstatin and cardioprotection studies in vitro
Rats were anaesthetized with pentobarbital sodium (50 mg/kg). Excised hearts were perfused retrogradely and under constant perfusion pressure (85 mm Hg) and instrumented as described previously.7 Hearts (n = 6/group) were subjected to 30 min regional ischaemia and 180 min reperfusion. Figure 1 depicts the treatment groups. Group A received no treatment, Group B received different concentrations of parstatin continuously for 15 min prior to coronary occlusion, Group C received an inhibitor (SB203580, PD98059, ODQ, L-NMA, or glibenclamide) 15 min before receiving parstatin, and Group D received the inhibitor (SB203580, PD98059, ODQ, L-NMA, or glibenclamide) alone. The rats in Groups E and F were treated with pertussis toxin (PTX) (10 µg/kg, ip) 48 h before isolating the heart.12,13 The heart was then treated with (Group E) or without (Group F) parstatin for 15 min prior to 30 min ischaemia. The measurements for post-ischaemic recovery of left ventricular developed pressure (LVDP) and coronary flow were taken at 60 min reperfusion and infarct size was determined after 180 min reperfusion. Coronary flow rate was determined by timed collection of the coronary effluent.
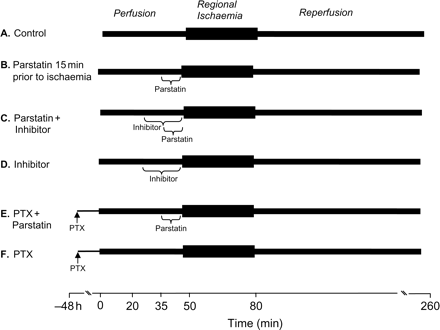
Isolated heart experimental protocols. All hearts were subjected to 30 min regional ischaemia after a 50 min stabilization period and followed by 180 min reperfusion. The control group (A) received no treatment. The parstatin group (B) was continuously perfused with different concentrations of parstatin 15 min before the onset of ischaemia. The inhibitor groups were continuously perfused with an inhibitor (SB203580, PD98059, ODQ, L-NMA, or glibenclamide) for 30 min before ischaemia with (C) or without (D) the addition of parstatin 15 min before ischaemia. (E and F) PTX was intraperitoneally administered to the rats 48 h before excision and perfusion of buffer with (E) or without (F) parstatin.
2.3 Constant-flow studies
To separate the effects of parstatin on coronary flow from those on contractile function, we repeated Groups A and B under conditions of constant flow. Coronary flow was adjusted to obtain a typical coronary perfusion pressure of 80–85 mmHg during the initial part of stabilization. Thereafter, this flow level (10 ± 1 mL/min/g) was maintained throughout the experiments. Hearts (n = 6/group) were treated with or without parstatin (1 µM) followed by 30 min regional ischaemia and 180 min reperfusion as described previously.14 Perfusion pressure was monitored throughout the study. Post-ischaemic coronary perfusion pressure and LVDP were measured at 60 min and infarct size was measured at 180 min reperfusion.
2.4 Infarct induction and measurement
The area at risk and infarct size were determined as described previously.11 The area at risk was 50 ± 3% of LV in all studies.
2.5 Immunoblot analysis
Left ventricular free wall tissue homogenates and immunoblot analysis were performed using methods described previously.11 Membranes were incubated with a 1:1000 dilution of the primary antibody [phospho-p38 mitogen-activated protein kinase (MAPK) or phospho-p44/42 MAPK (Thr202/Tyr204) antibodies] from Cell Signaling Technology (Danvers, MA, USA). An antibody against glyceraldehyde-3-phosphate dehydrogenase (Cell Signaling Technology) was used to standardize protein loading of each of the blots. A secondary antibody was then applied followed by enhanced chemiluminescence solution (Amersham Life Science, Arlington Heights, IL, USA) and exposed to film.
2.6 Microvessel vasodilation protocol
Coronary arterioles (50–180 µm internal diameters) were dissected from the LV free wall tissue of the isolated rat hearts after excision. Microvessel studies were performed by in vitro organ bath videomicroscopy, as described previously.15 The vessels were pre-constricted to ∼50% of its maximal diameter with endothelin-1 (10–100 nM). Endothelium-independent/-dependent relaxation to parstatin (1–1000 nM) was examined in the presence or absence of inhibitors: Nω-nitro-l-arginine methyl ester (L-NAME, 100 µM), 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ, 10 µM), and glibenclamide (10 µM). At the end of each dose–response curve, a single dose of papaverine (200 µM) was added to determine the maximal internal diameter for normalization of dilator responses. Denuded vessels were obtained by passing a hair through the vessel several times followed by injection of 0.3 mL of air.16 All values are expressed as a percentage of the maximum dilation to papaverine from the initially constricted diameter.
2.7 Statistical analysis
Data reported are mean ± SD. Statistical analysis was performed by the use of repeated measures ANOVA with the Greenhouse–Geisser adjustment used to correct for the inflated risk of a Type I error.17 If significant, the Mann–Whitney test was used as a second step to identify which groups were significantly different.17 Significance was set at P < 0.05.
3. Results
3.1 Parstatin and cardioprotection in vivo
We used parstatin, a synthetic peptide which is homologous to the N-terminal cleavage product of PAR1, to determine whether the cryptic peptide limited ischaemia–reperfusion injury in an in vivo rat model. We first performed a dose–response analysis to determine the optimal protective dose. Parstatin (1–25 µg/kg) was injected 15 min prior to ligating the left coronary artery. The principal endpoint of this study was infarct size, expressed as a percentage of the area at risk. Infarct size was 63 ± 2% of the area at risk in the control group. No significant change in infarct size was detected with the 1, 3, or 25 µg/kg doses of parstatin. However, a significant decrease in infarct size was detected with the 5–15 µg/kg doses with 10 µg/kg as the optimal dose (Figure 2A). These hearts had an infarct size of 46 ± 3% of the area at risk, which is a 26% reduction in infarct size compared with the control. Treatment with parstatin 15 min after ischaemia and 10 s after the onset of reperfusion still resulted in a 23% and 18% reduction in infarct size, respectively (Figure 2B).
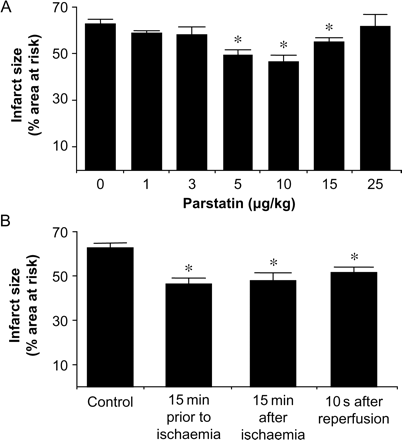
Analysis of the cardioprotective effects of parstatin in vivo. (A) Dose–response curve of parstatin. Rats were treated with either saline or parstatin (1–25 µg/kg) administered as an iv bolus 15 min prior to ischaemia. Infarct size was determined after 30 min regional ischaemia and 120 min reperfusion. (B) Phase of action of parstatin. Rats were treated with parstatin (10 µg/kg, iv) 15 min prior to ischaemia, 15 min after the onset of ischaemia, or 10 s after the onset of reperfusion. Data are mean ± SD, n = 6/group. *P < 0.05, treated vs. control.
No significant differences in heart rate and blood pressures were found between groups throughout the experiment. Mean arterial pressure decreased during ischaemia and reperfusion in all groups but there was no significant difference between groups (Table 1).
Haemodynamic values for parstatin in vivo dose–response studies
| Baseline | 15 min post-drug | 15 min ischaemia | 120 min reperfusion | |||||
|---|---|---|---|---|---|---|---|---|
| Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | |
| Drug-free control | 370 ± 15 | 124 ± 13 | n.a. | n.a. | 378 ± 15 | 99 ± 32 | 350 ± 13 | 82 ± 14 |
| Parstatin 1 µg/kg | 388 ± 15 | 125 ± 17 | 393 ± 6 | 120 ± 17 | 363 ± 35 | 81 ± 30 | 367 ± 21 | 79 ± 6 |
| Parstatin 3 µg/kg | 380 ± 42 | 132 ± 5 | 395 ± 35 | 136 ± 2 | 405 ± 35 | 128 ± 3 | 365 ± 64 | 110 ± 13 |
| Parstatin 5 µg/kg | 367 ± 15 | 132 ± 1 | 367 ± 12 | 128 ± 8 | 373 ± 15 | 115 ± 17 | 330 ± 10* | 78 ± 3 |
| Parstatin 10 µg/kg | 394 ± 15* | 115 ± 13 | 388 ± 8 | 107 ± 10 | 392 ± 28 | 98 ± 20 | 364 ± 33 | 76 ± 16 |
| Parstatin 15 µg/kg | 370 ± 27 | 133 ± 13 | 370 ± 27 | 125 ± 12 | 380 ± 17 | 120 ± 2 | 350 ± 17 | 88 ± 5 |
| Parstatin 25 µg/kg | 343 ± 32 | 118 ± 22 | 350 ± 27 | 119 ± 17 | 357 ± 29 | 115 ± 17 | 340 ± 35 | 85 ± 7 |
| Baseline | 15 min post-drug | 15 min ischaemia | 120 min reperfusion | |||||
|---|---|---|---|---|---|---|---|---|
| Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | |
| Drug-free control | 370 ± 15 | 124 ± 13 | n.a. | n.a. | 378 ± 15 | 99 ± 32 | 350 ± 13 | 82 ± 14 |
| Parstatin 1 µg/kg | 388 ± 15 | 125 ± 17 | 393 ± 6 | 120 ± 17 | 363 ± 35 | 81 ± 30 | 367 ± 21 | 79 ± 6 |
| Parstatin 3 µg/kg | 380 ± 42 | 132 ± 5 | 395 ± 35 | 136 ± 2 | 405 ± 35 | 128 ± 3 | 365 ± 64 | 110 ± 13 |
| Parstatin 5 µg/kg | 367 ± 15 | 132 ± 1 | 367 ± 12 | 128 ± 8 | 373 ± 15 | 115 ± 17 | 330 ± 10* | 78 ± 3 |
| Parstatin 10 µg/kg | 394 ± 15* | 115 ± 13 | 388 ± 8 | 107 ± 10 | 392 ± 28 | 98 ± 20 | 364 ± 33 | 76 ± 16 |
| Parstatin 15 µg/kg | 370 ± 27 | 133 ± 13 | 370 ± 27 | 125 ± 12 | 380 ± 17 | 120 ± 2 | 350 ± 17 | 88 ± 5 |
| Parstatin 25 µg/kg | 343 ± 32 | 118 ± 22 | 350 ± 27 | 119 ± 17 | 357 ± 29 | 115 ± 17 | 340 ± 35 | 85 ± 7 |
Values are mean ± SD, n = 6/group. Hearts subjected to 30 min regional ischaemia followed by 120 min reperfusion. MAP, mean arterial pressure.
*P < 0.05 vs. control group.
Haemodynamic values for parstatin in vivo dose–response studies
| Baseline | 15 min post-drug | 15 min ischaemia | 120 min reperfusion | |||||
|---|---|---|---|---|---|---|---|---|
| Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | |
| Drug-free control | 370 ± 15 | 124 ± 13 | n.a. | n.a. | 378 ± 15 | 99 ± 32 | 350 ± 13 | 82 ± 14 |
| Parstatin 1 µg/kg | 388 ± 15 | 125 ± 17 | 393 ± 6 | 120 ± 17 | 363 ± 35 | 81 ± 30 | 367 ± 21 | 79 ± 6 |
| Parstatin 3 µg/kg | 380 ± 42 | 132 ± 5 | 395 ± 35 | 136 ± 2 | 405 ± 35 | 128 ± 3 | 365 ± 64 | 110 ± 13 |
| Parstatin 5 µg/kg | 367 ± 15 | 132 ± 1 | 367 ± 12 | 128 ± 8 | 373 ± 15 | 115 ± 17 | 330 ± 10* | 78 ± 3 |
| Parstatin 10 µg/kg | 394 ± 15* | 115 ± 13 | 388 ± 8 | 107 ± 10 | 392 ± 28 | 98 ± 20 | 364 ± 33 | 76 ± 16 |
| Parstatin 15 µg/kg | 370 ± 27 | 133 ± 13 | 370 ± 27 | 125 ± 12 | 380 ± 17 | 120 ± 2 | 350 ± 17 | 88 ± 5 |
| Parstatin 25 µg/kg | 343 ± 32 | 118 ± 22 | 350 ± 27 | 119 ± 17 | 357 ± 29 | 115 ± 17 | 340 ± 35 | 85 ± 7 |
| Baseline | 15 min post-drug | 15 min ischaemia | 120 min reperfusion | |||||
|---|---|---|---|---|---|---|---|---|
| Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | Heart rate (bpm) | MAP (mmHg) | |
| Drug-free control | 370 ± 15 | 124 ± 13 | n.a. | n.a. | 378 ± 15 | 99 ± 32 | 350 ± 13 | 82 ± 14 |
| Parstatin 1 µg/kg | 388 ± 15 | 125 ± 17 | 393 ± 6 | 120 ± 17 | 363 ± 35 | 81 ± 30 | 367 ± 21 | 79 ± 6 |
| Parstatin 3 µg/kg | 380 ± 42 | 132 ± 5 | 395 ± 35 | 136 ± 2 | 405 ± 35 | 128 ± 3 | 365 ± 64 | 110 ± 13 |
| Parstatin 5 µg/kg | 367 ± 15 | 132 ± 1 | 367 ± 12 | 128 ± 8 | 373 ± 15 | 115 ± 17 | 330 ± 10* | 78 ± 3 |
| Parstatin 10 µg/kg | 394 ± 15* | 115 ± 13 | 388 ± 8 | 107 ± 10 | 392 ± 28 | 98 ± 20 | 364 ± 33 | 76 ± 16 |
| Parstatin 15 µg/kg | 370 ± 27 | 133 ± 13 | 370 ± 27 | 125 ± 12 | 380 ± 17 | 120 ± 2 | 350 ± 17 | 88 ± 5 |
| Parstatin 25 µg/kg | 343 ± 32 | 118 ± 22 | 350 ± 27 | 119 ± 17 | 357 ± 29 | 115 ± 17 | 340 ± 35 | 85 ± 7 |
Values are mean ± SD, n = 6/group. Hearts subjected to 30 min regional ischaemia followed by 120 min reperfusion. MAP, mean arterial pressure.
*P < 0.05 vs. control group.
3.2 Parstatin and cardioprotection in vitro
We then studied the effects of parstatin on ischaemia–reperfusion injury in an isolated heart model. We first performed a concentration–response analysis using 0.1, 1, or 10 µM to determine the optimal protective dose for the parstatin peptide. Control hearts produced an infarct size of 51 ± 3% of the area at risk (Figure 3A). Continuous administration of parstatin for 15 min immediately before ischaemia resulted in a concentration-dependent reduction of infarct size. However, 1 µM parstatin led to the largest reduction in infarct size (18 ± 3%), a 65% decrease. A picture representation of infarct in control vs. parstatin (1 µM)-treated hearts is shown in Figure 3C. Parstatin lost its efficacy when used at 10-fold higher or lower concentrations. Similarly, parstatin increased recovery of LVDP in a concentration-dependent manner. Again, the optimal concentration was 1 µM which produced a 23% recovery of LVDP (Figure 3B).
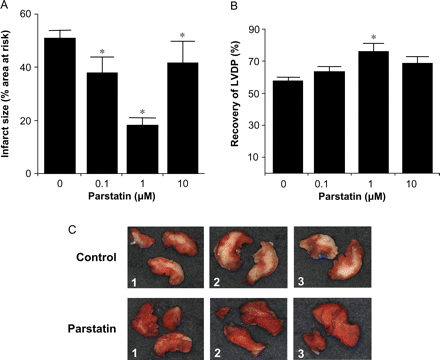
Cardioprotective effects of parstatin in vitro. Rat hearts were perfused with increasing concentrations of parstatin (0, 0.1, 1, and 10 µM) for 15 min prior to ischaemia. Infarct size and LVDP were determined after 30 min regional ischaemia and 180 min reperfusion. (A) Infarct size. (B) Recovery of LVDP. (C) Typical photographs of myocardial slices from three control and three parstatin-treated hearts. Infarcted areas are pale grey, whereas viable myocardium is dark red. Data are mean ± SD, n = 6/group. *P < 0.05, treated vs. control. LVDP, left ventricular developed pressure.
3.3 Role of Gi-protein-coupled receptors in parstatin-mediated cardioprotection in vitro
Rats were treated with PTX 48 h before ischaemia. In the presence of PTX, parstatin no longer is able to reduce infarct size or increase the recovery of LVDP after ischaemia–reperfusion injury (Figure 4A and B).
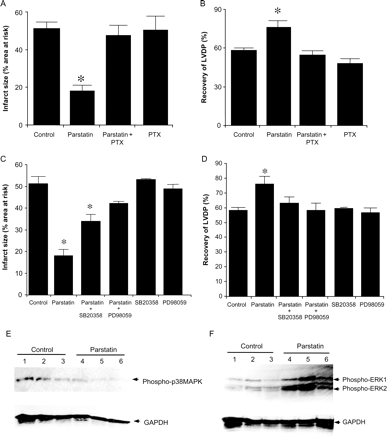
The cardioprotective effects of parstatin are dependent on Gi-protein signalling pathways. Inhibition of Gi-protein activation by PTX completely abolished the cardioprotective effects of parstatin. PTX was injected 48 h prior to ischaemia. Isolated heart were perfused +/− parstatin (1 µM) and subject to 30 min ischaemia and 180 min reperfusion. (A) Infarct size. (B) Recovery of LVDP. Inhibition of p38 MAPK or ERK1/2 negates the cardioprotective effects of parstatin. Isolated hearts were perfused with SB204580 (1 µM) or PD98059 (10 µM) with or without parstatin (1 µM). (C) Infarct size. (D) Recovery of LVDP. Data are mean ± SD, n = 6/group. *P < 0.05, treated vs. control. LVDP, left ventricular developed pressure. Parstatin does not increase activation of p38 MAPK but does increases activation of ERK1/2 after 5 min reperfusion as measured by phosphorylation of p38 MAPK and ERK1/2. Rat hearts were perfused with or without parstatin (1 µM) for 15 min before regional ischaemia. The free wall of the LV was harvested for protein extraction after 5 min reperfusion. Immunoblot for phosphorylated p38 MAPK (E) and phosphorylated ERK1/2 (F). GAPDH is the loading control. n = 3/group.
3.4 Role of MAPK in parstatin-mediated cardioprotection in vitro
Isolated hearts were pre-treating with SB203580 or PD98059, inhibitors of p38 MAPK and extracellular signal-regulated kinases 1/2 (ERK1/2), respectively, with or without parstatin treatment. p38 MAPK inhibition only partially blocks the infarct sparing effects of parstatin but yet completely abolishes the recovery of LVDP associated with parstatin treatment (Figure 4C and D). However, the protective effects of parstatin on both infarct size and recovery of LVDP were abolished by the pre-treatment of isolated hearts with PD98059, an ERK1/2 inhibitor (Figure 4C and D). We did not detect a difference in phosphorylated levels of p38 MAPK (Figure 4E) but found that the phosphorylation of ERK1/2 was greatly enhanced in parstatin-treated hearts at reperfusion when compared with control hearts (Figure 4F).
3.5 Role of and nitric oxide synthase in parstatin-mediated cardioprotection in vitro
Isolated hearts were perfused with the nitric oxide synthase (NOS) inhibitor, L-NMA (100 µM) with or without parstatin (1 µM) prior to ischaemia. L-NMA abolished the cardioprotective effect of parstatin but had no effect alone (Figure 5A and B).
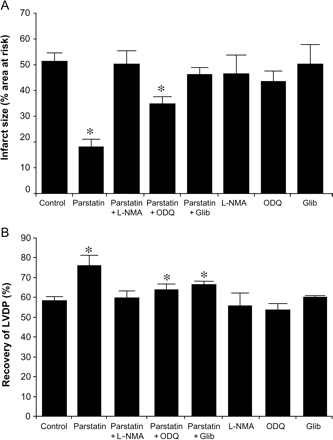
The cardioprotective effects of parstatin are dependent on NOS, sGC, and KATP channels. Inhibition of NOS (L-NMA, 100 µM), sGC (ODQ, 10 µM), or KATP channel (Glib, 1 µM). Isolated hearts were perfused with the inhibitors with or without parstatin (1 µM) before 30 min regional ischaemia and 180 min reperfusion. (A) Infarct size. (B) Recovery of LVDP. Data are mean ± SD, n = 6/group. *P < 0.05, treated vs. control. LVDP, left ventricular developed pressure.
3.6 Role of soluble guanylyl cyclase in parstatin-mediated cardioprotection in vitro
Isolated hearts were perfused with ODQ (10 µM), a potent and specific soluble guanylyl cyclase (sGC), inhibitor with or without parstatin (1 µM) prior to ischaemia. ODQ partially abolished reduction in infarct size caused by parstatin but had no effect alone (Figure 5A). ODQ completely reverses the recovery of LVDP resulting from parstatin treatment (Figure 5B).
3.7 Role of KATP channels in parstatin-mediated cardioprotection in vitro
Isolated hearts were perfused with the non-selective KATP channel blocker, glibenclamide, alone or with parstatin (1 µM) prior to ischaemia. Glibenclamide (3 µM) diminished the cardioprotective effect of parstatin (Figure 5A and B). Glibenclamide alone had no effect on infarct size or recovery of LVDP.
3.8 Role of vasodilation in parstatin-mediated cardioprotection in vitro
At an optimal concentration of 1 µM, we found that parstatin did not have inotropic or chronotropic effects before ischaemia (Table 2); however, in addition to preserving contractility during ischaemia–reperfusion, it also increased coronary flow during and after ischaemia (Figure 6C and Table 2).
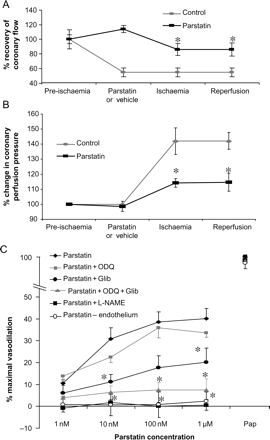
Coronary response to parstatin during ischaemia and reperfusion. (A) Parstatin increases coronary flow during ischaemia and reperfusion. Administration of parstatin (1 µM) 15 min prior to ischaemia and reperfusion resulted in an increase in coronary flow when compared with control values. (B) Parstatin decreases perfusion pressure in isolated rat hearts during ischaemia and reperfusion. Isolated rat hearts were perfused with or without parstatin (1 µM) for 15 min prior to regional ischaemia and reperfusion. Perfusion pressure was monitored throughout the procedure. Average pressures from pre-ischaemia, ischaemia, and post-ischaemia were compared. Data are mean ± SD, n = 6/group. *P < 0.05, parstatin vs. control. (C) Parstatin causes vasodilation in rat coronary arterioles that were pre-constricted with endothelin-1. This vasodilation is completely abolished by the pre-treatment with L-NAME and partially abolished with ODQ or glibenclamide. In addition, denuding the vessels also abolish the vasodilatory effects of parstatin. Maximal dilation was achieved with papaverine (Pap). Data are mean ± SD, n = 5/group. *P < 0.05, treated vs. control.
Haemodynamic values for parstatin-isolated heart dose–response studies
| Before drug administration | After drug administration | 30 min regional ischaemia | 60 min reperfusion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Constant pressure | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 256 ± 27 | 6.9 ± 1.3 | 141 ± 14 | n.a. | n.a. | n.a. | 225 ± 54 | 3.1 ± 0.6 | 83 ± 13 | 221 ± 18 | 3.9 ± 0.9 | 81 ± 4 |
| Parstatin 0.1 µM | 276 ± 26 | 5.8 ± 0.4 | 120 ± 5* | 250 ± 21 | 5.9 ± 0.6 | 106 ± 3 | 231 ± 45 | 3.5 ± 0.8 | 68 ± 7 | 241 ± 48 | 4 ± 0.7 | 76 ± 5 |
| Parstatin 1 µM | 265 ± 48 | 7 ± 0.6 | 125 ± 13 | 257 ± 47 | 7.9 ± 0.6 | 118 ± 15 | 244 ± 67 | 5.7 ± 0.9* | 81 ± 9 | 257 ± 39* | 5.9 ± 0.6* | 95 ± 12* |
| Parstatin 10 µM | 275 ± 4 | 6 ± 0 | 118 ± 10 | 255 ± 8 | 4.9 ± 0.3 | 107 ± 9 | 238 ± 15 | 3.2 ± 0.3 | 80 ± 5 | 218 ± 40 | 3 ± 0.1 | 82 ± 10 |
| Constant flow | Heart rate (bpm) | Coronary perfusion pressure (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary Perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 228 ± 27 | 80 ± 4 | 127 ± 12 | n.a. | n.a | n.a. | 216 ± 45 | 42 ± 9 | 93 ± 15 | 216 ± 27 | 43 ± 6% | 72 ± 13 |
| Parstatin 1 µM | 239 ± 16 | 87 ± 3 | 122 ± 6 | 240 ± 14 | -2 ± 3 | 125 ± 7 | 236 ± 16 | 14 ± 3* | 79 ± 17 | 252 ± 45 | 15 ± 6* | 92 ± 7* |
| Before drug administration | After drug administration | 30 min regional ischaemia | 60 min reperfusion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Constant pressure | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 256 ± 27 | 6.9 ± 1.3 | 141 ± 14 | n.a. | n.a. | n.a. | 225 ± 54 | 3.1 ± 0.6 | 83 ± 13 | 221 ± 18 | 3.9 ± 0.9 | 81 ± 4 |
| Parstatin 0.1 µM | 276 ± 26 | 5.8 ± 0.4 | 120 ± 5* | 250 ± 21 | 5.9 ± 0.6 | 106 ± 3 | 231 ± 45 | 3.5 ± 0.8 | 68 ± 7 | 241 ± 48 | 4 ± 0.7 | 76 ± 5 |
| Parstatin 1 µM | 265 ± 48 | 7 ± 0.6 | 125 ± 13 | 257 ± 47 | 7.9 ± 0.6 | 118 ± 15 | 244 ± 67 | 5.7 ± 0.9* | 81 ± 9 | 257 ± 39* | 5.9 ± 0.6* | 95 ± 12* |
| Parstatin 10 µM | 275 ± 4 | 6 ± 0 | 118 ± 10 | 255 ± 8 | 4.9 ± 0.3 | 107 ± 9 | 238 ± 15 | 3.2 ± 0.3 | 80 ± 5 | 218 ± 40 | 3 ± 0.1 | 82 ± 10 |
| Constant flow | Heart rate (bpm) | Coronary perfusion pressure (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary Perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 228 ± 27 | 80 ± 4 | 127 ± 12 | n.a. | n.a | n.a. | 216 ± 45 | 42 ± 9 | 93 ± 15 | 216 ± 27 | 43 ± 6% | 72 ± 13 |
| Parstatin 1 µM | 239 ± 16 | 87 ± 3 | 122 ± 6 | 240 ± 14 | -2 ± 3 | 125 ± 7 | 236 ± 16 | 14 ± 3* | 79 ± 17 | 252 ± 45 | 15 ± 6* | 92 ± 7* |
Values are mean ± SD, n = 6/group. Hearts subjected to 30 min regional ischaemia followed by 60 min reperfusion.
*P < 0.05 vs. control group.
Haemodynamic values for parstatin-isolated heart dose–response studies
| Before drug administration | After drug administration | 30 min regional ischaemia | 60 min reperfusion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Constant pressure | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 256 ± 27 | 6.9 ± 1.3 | 141 ± 14 | n.a. | n.a. | n.a. | 225 ± 54 | 3.1 ± 0.6 | 83 ± 13 | 221 ± 18 | 3.9 ± 0.9 | 81 ± 4 |
| Parstatin 0.1 µM | 276 ± 26 | 5.8 ± 0.4 | 120 ± 5* | 250 ± 21 | 5.9 ± 0.6 | 106 ± 3 | 231 ± 45 | 3.5 ± 0.8 | 68 ± 7 | 241 ± 48 | 4 ± 0.7 | 76 ± 5 |
| Parstatin 1 µM | 265 ± 48 | 7 ± 0.6 | 125 ± 13 | 257 ± 47 | 7.9 ± 0.6 | 118 ± 15 | 244 ± 67 | 5.7 ± 0.9* | 81 ± 9 | 257 ± 39* | 5.9 ± 0.6* | 95 ± 12* |
| Parstatin 10 µM | 275 ± 4 | 6 ± 0 | 118 ± 10 | 255 ± 8 | 4.9 ± 0.3 | 107 ± 9 | 238 ± 15 | 3.2 ± 0.3 | 80 ± 5 | 218 ± 40 | 3 ± 0.1 | 82 ± 10 |
| Constant flow | Heart rate (bpm) | Coronary perfusion pressure (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary Perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 228 ± 27 | 80 ± 4 | 127 ± 12 | n.a. | n.a | n.a. | 216 ± 45 | 42 ± 9 | 93 ± 15 | 216 ± 27 | 43 ± 6% | 72 ± 13 |
| Parstatin 1 µM | 239 ± 16 | 87 ± 3 | 122 ± 6 | 240 ± 14 | -2 ± 3 | 125 ± 7 | 236 ± 16 | 14 ± 3* | 79 ± 17 | 252 ± 45 | 15 ± 6* | 92 ± 7* |
| Before drug administration | After drug administration | 30 min regional ischaemia | 60 min reperfusion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Constant pressure | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Coronary flow rate (mL/min/g) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 256 ± 27 | 6.9 ± 1.3 | 141 ± 14 | n.a. | n.a. | n.a. | 225 ± 54 | 3.1 ± 0.6 | 83 ± 13 | 221 ± 18 | 3.9 ± 0.9 | 81 ± 4 |
| Parstatin 0.1 µM | 276 ± 26 | 5.8 ± 0.4 | 120 ± 5* | 250 ± 21 | 5.9 ± 0.6 | 106 ± 3 | 231 ± 45 | 3.5 ± 0.8 | 68 ± 7 | 241 ± 48 | 4 ± 0.7 | 76 ± 5 |
| Parstatin 1 µM | 265 ± 48 | 7 ± 0.6 | 125 ± 13 | 257 ± 47 | 7.9 ± 0.6 | 118 ± 15 | 244 ± 67 | 5.7 ± 0.9* | 81 ± 9 | 257 ± 39* | 5.9 ± 0.6* | 95 ± 12* |
| Parstatin 10 µM | 275 ± 4 | 6 ± 0 | 118 ± 10 | 255 ± 8 | 4.9 ± 0.3 | 107 ± 9 | 238 ± 15 | 3.2 ± 0.3 | 80 ± 5 | 218 ± 40 | 3 ± 0.1 | 82 ± 10 |
| Constant flow | Heart rate (bpm) | Coronary perfusion pressure (mL/min/g) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary Perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) | Heart rate (bpm) | Change in coronary perfusion pressure from baseline (%) | Left ventricular developed pressure (mmHg) |
| Drug-free control | 228 ± 27 | 80 ± 4 | 127 ± 12 | n.a. | n.a | n.a. | 216 ± 45 | 42 ± 9 | 93 ± 15 | 216 ± 27 | 43 ± 6% | 72 ± 13 |
| Parstatin 1 µM | 239 ± 16 | 87 ± 3 | 122 ± 6 | 240 ± 14 | -2 ± 3 | 125 ± 7 | 236 ± 16 | 14 ± 3* | 79 ± 17 | 252 ± 45 | 15 ± 6* | 92 ± 7* |
Values are mean ± SD, n = 6/group. Hearts subjected to 30 min regional ischaemia followed by 60 min reperfusion.
*P < 0.05 vs. control group.
To investigate whether the increase in coronary flow was due to parstatin-specific vasoactivity, isolated rat hearts (n = 6/group) were perfused under constant-flow (10 mL/min/g) conditions with or without parstatin (1 µM) and subject to 30 min regional ischaemia and 180 min reperfusion. Coronary perfusion pressure which is an indicator of vascular reactivity in this system was monitored.
The ischaemia–reperfusion insult in control hearts increased the coronary perfusion pressure by 142 ± 3% during regional ischaemia and reperfusion when compared with pre-ischaemic values (Table 2 and Figure 6B). Parstatin limited this increase to 114 ± 4% pre-ischaemic values. Furthermore, under constant-flow conditions, parstatin limited the infarct size to 26 ± 2% area at risk (vs. 43 ± 2% control; data not shown) and increased the recovery of post-ischaemic function to 75 ± 6% (vs. 56 ± 5% control) of pre-ischaemic LVDP (data not shown).
3.9 Parstatin causes endothelium-dependent vasodilation in isolated coronary arterioles
Coronary arterioles were dissected from rat ventricular myocardium and prepared for video microscopy assessment of diameter as described in Section 2. Graded doses of parstatin resulted in a modest and incremental but potent dilation with an ED50 in the nanomolar range. Parstatin-induced vasodilation was abolished by pre-treatment of the vessels with L-NAME and reduced significantly with glibenclamide. ODQ did not significantly reduce vasodilation but the combination of ODQ and glibenclamide resulted in an additive effect to prevent parstatin-mediated vasodilation. Furthermore, parstatin does not vasodilate endothelium-denuded vessels (Figure 6C).
4. Discussion
There are several important new findings in the present study. First, brief exposure to parstatin preconditioned the rat heart and decreased infarct size, improved the recovery of LVDP, and increased coronary perfusion. More importantly, parstatin administration after the onset of reperfusion was still cardioprotective. This underscores the translational potential of this compound. Secondly, the co-administration of an ERK1/2, p38 MAPK, NOS, sGC, and KATP inhibitors diminished the beneficial effects of parstatin-induced preconditioning. Thirdly, in normal coronary arterioles, parstatin induces a potent vasodilation through an endothelium-dependent NO and KATP mechanisms.
Preconditioning puts the heart into a state of self-preservation. Preconditioning is triggered by either brief cycles of ischaemia or by exogenous compounds (i.e. adenosine and opioids),18 which typically activate Gi-protein-coupled surface receptors, AMP-activated kinase,19 and 3′-phosphoinositide-dependent kinase-120 to set off a complex pathway which ultimately results in cell survival. Gi-protein activation leads to the activation of members of the MAPK family including p38 MAPK and ERK1/2; important mediators of cardioprotection.21–24 ERK1/2 is known to activate NOS25–28 and the production of NO subsequently targets sGC which results in the conversion of guanosine-5′-triphophate to the intracellular second messenger cyclic guanosine-3′,5′-monophosphate (cGMP). KATP channels are opened in a cGMP-dependent manner.27,29,30 These steps are thought to occur prior to the index ischaemia and are able to put the heart into its preconditioned state. Opening of the KATP channels generates reactive oxygen species which are thought to be secondary messengers for kinase activation.31 Therefore, by the time of reperfusion prosurvival kinases including ERK1/2 are quickly activated32 and protect the heart against reperfusion injury.33,34 In addition, activation of sarcolemmal KATP channels shortens action membrane potential duration and decreases intracellular Ca2+ loading, which also leads to cardioprotection.20,35
Our data demonstrates that parstatin acts through a Gi-protein-mediated pathway to induce cardioprotection by modulating both the trigger and mediator phases of preconditioning. Blockade of p38 MAPK, ERK1/2, NOS, and KATP channels prior to ischaemia abolishes the infarct sparing and recovery of LVDP effects of parstatin. This implicates parstatin's role in mediating preconditioning through previously delineated mediators.
However, we did not detect equal contribution to parstatin-mediated cardioprotection by all factors examined. Only ERK1/2 and NOS inhibition completely reversed parstatin-mediated cardioprotection. p38 MAPK inhibition resulted in the partial reversal of parstatin cardioprotection. Furthermore, parstatin treatment before ischaemia is able to increase the phosphorylation of ERK1/2 but not p38 MAPK at reperfusion. These findings indicate that either p38 MAPK is upstream of ERK1/2 or in a divergent pathway. p38 MAPK may be an early trigger whereas ERK1/2 is most likely a trigger and mediator of parstatin-induced cardioprotection.
Furthermore, sGC and KATP channel inhibition also resulted in the partial reversal of cardioprotection by parstatin. Thus, parstatin-mediated cardioprotection appears to be mediated in part by a pathway independent of sGC activity. Previously, it had been shown that the KATP channel is regulated by a cGMP-dependent protein kinase signalling pathway.30 However, it has also been shown that KATP channels can be opened in an sGC-independent manner relying instead on ERK1/2 phosphorylation.36,37 In this study, although the parstatin-mediated cardioprotection is stimulated by a cGMP-KATP channel-dependent process, the complete action of parstatin does not require it.
On the other hand, the complete action of parstatin does require NOS and presumably NO signalling. NO can modify proteins involved in the signalling mechanism by chemical reaction such as S-nitrosylation other than by means of activation of sGC.38S-Nitrosylation has also been shown to be involved in myocardial protection from ischaemia–reperfusion injury.39 Indeed, our data suggests that parstatin-mediated cardioprotection is mediated by both NO activation of sGC and NO activation of other mediating proteins. The resultant S-nitrosylation effects of parstatin are yet to be determined.
Nitric oxide, ERK1/2, and KATP channels appear to serve some common physiological functions, such as vasodilation and cardioprotection. Indeed, the signalling pathways responsible for coronary artery vasodilation are somewhat parallel to the signalling pathways responsible for preconditioning and cardioprotection. During ischaemia and reperfusion in the isolated heart, parstatin increases coronary flow under constant coronary pressure conditions. Typically, coronary flow is tightly coupled to oxygen demand. In non-diseased coronary vessels, whenever cardiac activity and oxygen consumption increases, there is an increase in coronary blood flow that is nearly proportionate to the increase in oxygen consumption. In innervated hearts, increased metabolic activity from increased heart rate and/or contractility triggers sympathetic activation which results in coronary vasodilation and increased coronary flow. The advantage of the isolated heart model is the examination of contractility, heart rate, and vascular effects without the neuronal and hormonal complications of an intact animal model. We confirmed the vasodilatory effects of parstatin in the isolated heart under constant-flow conditions. In this model, higher perfusion pressure correlates with vasoconstriction and lower perfusion pressure correlates with vasodilation. Parstatin reduced coronary perfusion pressure during regional ischaemia and reperfusion. Furthermore, parstatin salvaged the ischaemic myocardium and restores contractile function under both constant perfusion and constant-flow conditions, suggesting that parstatin exerts its cardioprotective functions by acting on both myocardial myocytes and vasculature.
Since parstatin increased coronary flow during ischaemia and reperfusion in rat hearts, we reasoned that parstatin was mediating vasodilation of the coronary microvasculature. Our data also demonstrates that parstatin mediates vasodilation in an endothelium and NO-dependent mechanism. Paralleling our cardioprotection results, sGC and KATP channel inhibition do not completely abolish parstatin-mediated vasodilation. Moreover, sGC inhibition does not significantly reverse parstatin's vasodilatory effects. sGC and KATP channel inhibition are additive in abolishing parstatin-mediated vasodilation. In this case, NO may be vasodilating in a cGMP-KATP channel-dependent and -independent manner. NO is also known to mediate vasodilation by activating other potassium channels.40,41 In conclusion, parstatin mediates endothelium-dependent vasodilation in coronary arterioles through a mechanism that involves NO and KATP channels.
In conclusion, we have demonstrated that parstatin, the N-terminal cleavage product of PAR1, is an effective agent for cardioprotection during ischaemia and reperfusion of the rat myocardium. We have also shown that parstatin causes vasodilation in isolated rat coronary arterioles. Both cardioprotection and vasodilatory properties of parstatin is largely dependent on NOS to a lesser extent on sGC and KATP channels. Parstatin is a novel and intriguing activator of cardioprotection and vasodilation. Parstatin manifests its effects in part through known mediators of cardioprotection and vasodilation, suggesting that it is acting through multiple pathways to exert its effects. These other pathways may include S-nitrosylation or other potassium channels.
Collectively, the results of these studies in rat hearts and coronary vessels strongly suggest that parstatin serves a protective role during ischaemia–reperfusion and induces coronary vasodilation. Since the cardioprotective effects of the parstatin occurred in the absence of haemodynamic changes, there is an exciting opportunity to develop parstatin to protect against myocardial and microvascular injury in the clinical setting.
Conflict of interest: none declared.
Funding
This study was supported in part by National Institutes of Health Grant HL54075 to J.E.B. and Advancing Healthier Wisconsin Grant 5520119-9520094 awarded to M.E.W. J.L.S. was supported by National Institutes of Health Training Grant HL07792 (awarded to the Medical College of Wisconsin). This work was also supported by the University of Patras Research Committee (Program ‘k. Karatheodoris 2005’).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}