





Abstract
Objectives The role of A2 noradrenergic neurons in regulating cardiovascular homeostasis chronically is poorly understood. We aimed to genetically target A2 neurons and induce expression of a potassium channel to reduce their electrical excitability and study how this impacts on long-term blood pressure control.
Methods We used a lentiviral vector with PRSx8 promoter for targeting noradrenergic neurons to express a human inwardly rectifying potassium channel, hKir2.1. The dorsal vagal complex containing the A2 cell group was microinjected with the PRSx8-hKir2.1 lentivirus in both normotensive Wistar rats and spontaneously hypertensive rats fitted with radio telemetry devices.
Results In Wistar rats expression of hKir2.1 increased lability of arterial pressure between 7 to 21 days post-injection with mean arterial pressure not increasing significantly until day 21 (+11±1 mmHg; p<0.001; dark phase). Urine output and water intake were both decreased. In contrast, in spontaneously hypertensive rats not only the lability of arterial pressure but also the mean arterial pressure increased by day 7 and persisted during the 21 day recording period (+13±1 mmHg; p<0.001 at day 21). In contrast to Wistar rats, body fluid homeostasis was un-affected in hypertensive rats. Neither cardiac baroreceptor reflex gain nor heart rate variability changed in either rat strain. Plasma osmolality levels were also unaffected.
Conclusions Our data indicate a role for A2 neurons in the chronic regulation of arterial pressure independent of the cardiac baroreceptor reflex. The activity of A2 neurons may constitute an essential part of the central circuitry underpinning chronic regulation of arterial pressure in both, normo- and hypertensive rats.
Time for primary review 21 days
1 Introduction
The idea of a set-point controller in the brain regulating arterial pressure is not new [1] but remains hypothetical as the neural substrate has yet to be realised. With the recent debate on the role of arterial baroreceptors in the long term control of arterial pressure [2–5] it seems pertinent to focus on potential roles of defined groups of central neurons for chronic control of arterial pressure.
One such group are the A2 noradrenergic (NEergic) neurons in the nucleus of the solitary tract (NTS). Three decades ago it became apparent that A2 neurons may be important for blood pressure regulation and could be a key link in the mechanisms of action of central anti-hypertensive drugs, such as clonidine and α-methylnoradrenaline [6–9]. Several studies reported that in normotensive rats localised ablation of the NTS by electrolytic lesioning produced fulminating hypertension [10–12]. However, such lesions are not confined to A2 neurons. Relatively more selective destruction of A2 neurons, using the neurotoxin 6-hydroxydopamine (6-OHDA), de-stabilised arterial pressure by causing lability [12,13], without affecting mean arterial pressure (MAP) [14]. Moreover, Talman et al. (1980) reported that the cardiac baroreceptor reflex was attenuated after destroying A2 neurons with 6-OHDA [12], but the reflex depressor response to carotid sinus stretch was unaffected. However, whether A2 neurons are involved in mediating or modulating the baroreflex remains uncertain and the limited data appear contradictory [15,16]. The problem is further confounded by the fact that 6-OHDA is not selective: it is a general neurotoxin and destroys serotonin containing neurons projecting to the NTS [17]. Finally, notwithstanding the insights yielded from the aforementioned lesioning studies, any method that destroys neurons may lead to tissue necrosis, axonal sprouting and re-wiring, compromising the integrity of the neuronal networks [18–20] and making data interpretation difficult.
Our approach was to avoid some of these limitations and electrically “silence”, rather than destroy, A2 neurons. In NEergic neurons the transcription factor Phox2 is highly active. This feature is exploited by an artificial Phox2-dependent promoter [21], PRSx8, which we used to express a human inwardly rectifying potassium channel (hKir2.1) [22]. Using this approach, we have been able to assess the role of A2 neurons chronically by reducing their electrical excitability whilst monitoring cardiovascular parameters and body fluid homeostasis in freely moving normo- and pre-programmed genetically hypertensive rats. Our hypothesis was that reducing the excitability of A2 neurones (by virally mediated expression of Kir2.1) arterial pressure in both normotensive and spontaneously hypertensive rats will become elevated.
Part of this work was communicated in abstract form [23].
2 Materials and methods
2.1 Viral vector construction and validation
Lentiviral vector (LVV) construction was based on a previous study24. Details for production and purification are described in Supplement 1. Validation of transduction efficacy and transgene expression was assessed using (i) enhanced green fluorescent protein (eGFP) fluorescence and immunocytochemistry (Fig. 1A–D), (ii) detection of hKir2.1 and eGFP mRNAs by reverse transcription polymerase chain reaction (RT-PCR; Fig. 1E), and (iii) by recording membrane potential in whole-cell patch clamp experiments in transiently transfected (Superfect, QIAGEN) PC12 cells (Fig. 1F). Details of these experimental procedures are described in Supplement 1.
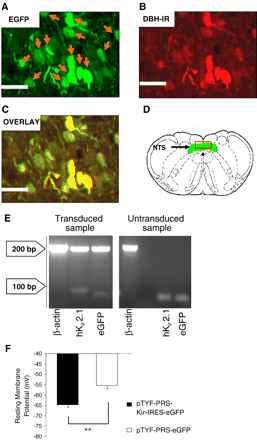
Validation of hKir2.1 expression in vivo and in vitro. Microinjection of LVV-PRS-Kir-IRES-eGFP into the NTS resulted in eGFP-expressing neurons (A) which stained positive for the NEergic marker DBH (B). The overlay (C) shows that the majority of DBH immunoreactivity (IR) co-localised with eGFP IR. Scale bar 40 μm. D shows a schematic transverse section of the caudal medulla (DVM, green) with the red box highlighting the region corresponding to panels A–C. Agarose electrophoresis of RT-PCR products (E) confirmed the expression of hKir2.1 and eGFP only in LVV-PRS-Kir-IRES-eGFP transduced NTS but not in naïve tissue. The membrane potential in PC12 cells transfected with pTYF-PRS-Kir-IRES-eGFP (PRSx8 promoter cloned in antisense direction) was significantly more negative than in pTYF-PRS-eGFP transfected controls (−64.7±−1.2 mV vs. −55.3±−1.6 mV, p<0.01) There was no significant difference in membrane potential in PC12 cells transfected with the sense and antisense pTYF-PRS-Kir-IRES-eGFP (data not shown).
2.2 Radio-telemetry measurement of cardiovascular autonomic activity
All procedures were in accordance with the Home Office Animal (Scientific Procedures) Act 1986. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Male Wistar rats (WR) and spontaneously hypertensive rats (SHR) weighing between 280–300 g (12–14 weeks old) were housed for the duration of the study (one week prior to and four weeks after viral vector microinjection) individually in metabolic cages (Tecniplast no. 3700 M071, Gazzada, Italy). WR were used as a normotensive comparator as each strain had its own control. In addition, based on their undefined genetic lineage to SHR, WR would add robustness to any differences in cardiovascular responses observed. Rats were supplied with normal rat chow and drinking water ad libitum, and kept on a 12 h light–12 h dark cycle in a sound proofed, temperature and humidity controlled room. Water drunk and urine produced were measured daily. A radio-telemetry system (Data Sciences International, St Paul, MN, USA) was used for recording arterial pressure [24] and a computer-based data acquisition system was used to acquire, display, store and analyse the data as described recently [25]. A blood pressure radio transmitter (TA11PA-C40) was implanted 7 days before recordings began, as before [24]. During the two days preceding microinjection of vectors (see below) and on the 7th, 14th and 21st day after microinjection, twelve 5 min periods of pulsatile arterial pressure were collected (on the hour) throughout both light and dark phases (see Supplement 2). For the power spectral analysis of arterial pressure, heart rate (HR) and respiratory rate data and spontaneous baroreceptor reflex gain (sBRG), see Supplement 1.
2.3 Injection of LVV in the NTS
Animals with pre-implanted transmitters were re-anaesthetized 9 days after implantation of radio-transmitter with ketamine (60 mg kg−1) and medetomidine (250 μg kg−1) mixture. They were placed in a stereotaxic head holder and through a midline incision in the dorsal neck, the caudal dorsomedial medulla was exposed. Bilateral microinjection of LVV (108 ifu min−1; 500 nl injections per side) were made at the level of the calamus scriptorius and 250 μm rostral and caudal to it, 300–500 μm from the midline and 500 μm ventral to the dorsal surface of the medulla. Two groups of both SHR and WR were injected (see Supplement 2). The first group of either strain received microinjections of LVV-PRS-Kir-IRES-eGFP, while the second group acted as control for LVV transfection and received LVV-PRS-eGFP. The wound was sutured, cleaned and treated with antiseptic powder and anaesthesia reversed with a subcutaneous injection of atipamezole (1 mg kg−1). Afterwards animals were returned to their cages for recovery. Data were analysed in which the NTS was demonstrated to have been transduced successfully with post-hoc histology. All injection sites were within the dorsal vagal complex centred around the caudal NTS that corresponded rostro-caudally to the level of area postrema to the commissural NTS.
2.4 Data analysis
Data for hKir2.1- and eGFP-expressing groups are expressed as means±S.E.M. To evaluate differences in A2 and eGFP expressing neurons and time-dependent changes of cardiovascular variables following expression of hKir2.1 within the A2 neurons, we used repeated-measures ANOVA and the Bonferroni test for multiple comparisons of cardiovascular variables across time and between different groups. Data were taken as significant at p<0.05.
3 Results
3.1 Validation of expression and functional effect of LVV-PRS-Kir-IRES-eGFP
We confirmed immunohistochemically that A2 neurons expressed eGFP in the LVV-PRS-Kir-IRES-eGFP transduced NTS (Fig. 1A–D). At least 30% DBH-positive neurons also expressed eGFP in the NTS. We also noted that in LVV-PRS-Kir-IRES-eGFP transduced rats there were eGFP-positive neurons in the dorsal vagal motor nucleus (DVM). This was inevitable as dendrites of these neurons project into the NTS and we previously found PRSx8 activity in DVM neurons [26]. We compared cell counts of eGFP expressing and immunolabeled neurones between WR and SHR. No difference was found in the number of eGFP expressing neurons between WR and SHR (p=0.44; ANOVA, based on counts from 6 animals) suggesting that the efficacy of transduction was similar between rat strains (please see Supplement 3). Moreover, we counted the number of DBH immunostained noradrenergic neurons in SHR and WR and again, found no difference (p=0.5; ANOVA, based on counts from 6 animals; please see Supplement 1). The rostro-caudal extent of the transduced area coincided with 12.80–14.60 mm relative to bregma and included parts of the commissural, dorsomedial and central sub-nuclei of the NTS. Further, we found strong signals for PCR products of both eGFP and hKir2.1 in NTS tissue microdissected from the NTS from transduced but not from naïve rats (Fig. 1E).
eGFP-expressing PC12 cells transfected with either pTYF-PRS-eGFP or pTYF-PRS-Kir-IRES-eGFP shuttle plasmids were identified using conventional epifluorescence and patch clamped. In pTYF-PRS-eGFP-transfected control cells, resting membrane potential was −55.3±−1.6 mV (n=15 cells). In contrast, the membrane potential of cells transfected with pTYF-PRS-Kir-IRES-eGFP was significantly more negative (i.e. −64.7±−1.2 mV; n=16 cells, p<0.01). These data demonstrate that expression of hKir2.1 can hyperpolarize membrane potential by approximately 9 mV (Fig. 1F), which is consistent with previous studies where hKir2.1 was used to depress neuronal excitability [22,27]. From experiments in slice cultures we found that the basic membrane characteristics of A2 neurones in WR and SHR (e.g. resting membrane potential and input resistance) are not different (please see Supplement 4). It follows that our construct may be expected to have a comparable “silencing” effect on A2 cell group in both rat strains.
3.2 Chronic expression of hKir2.1 in the A2 cell group of WR and SHR rats
Baseline MAP for WR and SHR during the light phase was 96±2.6 and 144±2.5 mmHg respectively (Fig. 2A). Control levels of HR and sBRG were 350±6 bpm and 1.13±0.1 bpm ms−1 for WR, and 364±2 bpm and 0.78±0.7 bpm ms−1 for SHR, respectively. Mean values during the dark phase were not significantly different from light phase data within each group (Fig. 3). Body weights of both groups of animals (hKir2.1 and eGFP) were not different significantly prior to viral injections. However, both did increase at 21 days (eGFP: from 287 to 364 g; Kir: from 293 to 444 g; n=6, p<0.001). The gain in body weight in the Kir transduced rats was greater than in the eGFP control group (p<0.01).
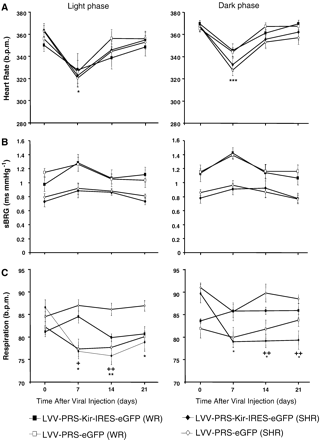
Effects of chronic overexpression of hKir2.1 in A2 NEergic neurons on HR, sBRG, and respiration in WR and SHR. A. In neither rat strain transduced with LVV-PRS-Kir-IRES-eGFP during either the light or dark phases during the 21 day observation period was there a change in HR (except on day 7 in both strains; see Results section). B. The sBRG in SHR was lower than in WR but unaffected by LVV-PRS-Kir-IRES-eGFP transduction in both strains. C. Expression of hKir2.1, but not eGFP, in the A2 cell group resulted in a significant decrease in respiratory rate in SHR but not WR. Square and diamond symbols indicate WR and SHR data. Open and closed data refer to LVV-PRS-eGFP and LVV-PRS-Kir-IRES-eGFP viruses.
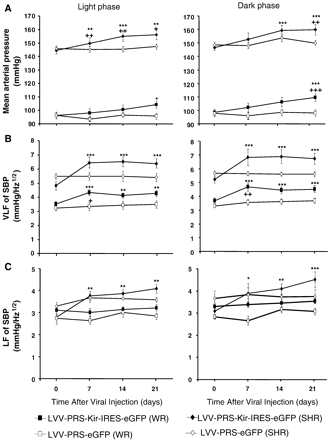
Effects of chronic overexpression of hKir2.1 in A2 NEergic neurons on MAP, LF and VLF of SBP in WR and SHR. A. A significant time-dependent increase in MAP was observed in hKir2.1-transduced WR (n=6) and SHR (n=6). B: increases in MAP occurred two weeks earlier in SHR. There was no significant change in MAP in the eGFP transduced control groups of both rat strains (n=4 in each case).B. VLF of SBP in hKir2.1-transduced normotensive WR and SHR significantly increased from day 7 post-injection (A–B), this was most evident during the dark phase (right). C. In contrast, only hKir2.1-transduced SHR showed a significant increase in LF of SBP (C), which was most evident during the dark phase (right panel). There was no significant change in VLF of SBP and LF of SBP in the eGFP transduced groups of both rat strains. Square and diamond symbols indicate WR and SHR data. Open and closed data refer to LVV-PRS-eGFP and LVV-PRS-Kir-IRES-eGFP viruses.
3.2.1 MAP changes
In the WR, the MAP remained unchanged for the first two weeks post transduction but by day 21 it deviated significantly from eGFP-only expressing control rats (+11 mmHg, p<0.001; Fig. 2A). In contrast, in SHR expression of hKir2.1 resulted in a significant elevation of arterial pressure by day 7 and reached +13 mmHg by day 21. These changes were apparent during both light and dark phases (Fig. 2A). There was no significant change in MAP in the eGFP-transduced rats of both strains at any time point during either light or dark phase.
3.2.2 Spectral analysis of systolic blood pressure (SBP)
In WR, very low frequency (VLF) of SBP increased significantly from day 7 post-viral transduction during both light and dark phases, suggestive of an enhanced lability of arterial pressure (Fig. 2B). This was further supported by a significant increase in the standard deviation of arterial pressure (light phase: from 5.12 to 9.22; p<0.001). VLF also increased in SHR but to a greater extent (SHR Δ 1.7±0.2 vs. WR Δ 0.6±0.1 mmHg Hz−1 1/2, p<0.001; Fig. 2B). Moreover, the standard deviation of MAP increased from 4.42 to 9.17 in the SHR (light phase; p<0.001). In contrast to WR, SHR also exhibited a sustained and significant increase in the low frequency (LF) spectra from day 7 post-transduction (Fig. 2C) suggestive of elevated sympathetic vasomotor tone. eGFP transduced controls in both rat strains showed no significant change in VLF, LF spectra or standard deviation of MAP throughout the observation period (Fig. 2B–C).
3.2.3 HR and cardiac baroreceptor reflex gain
In both rats strains transduced with either of the constructs there was a transient decrease in HR at day 7, which then recovered to the pre-injection level (Fig. 3A). Given that this transient effect was not related to expression of hKir2.1, it could be related to the surgery. In neither rat strain transduced with hKir2.1 during either the light or dark phases during the 21 day observation period was there a change in the sBRG compared to basal values (Fig. 3A). There were also no changes in these variables in the eGFP-transduced control rats during both light and dark phases, which were not different to the hKir2.1 transduced animals (Fig. 3A–B). Finally, there were no changes in the high frequency (HF) of pulse interval and LF/HF ratios (Fig. 4).
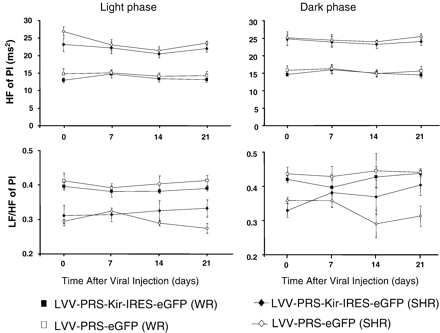
HR variability after hKir2.1 transduction of A2 neurons. There was no significant change in cardiac parasympathetic (HF of PI) and cardiac sympathetic tone (LF/HF of PI) in hKir2.1-transduced WR and SHR during the light phase (A and B) and dark phase (C and D). Similarly, eGFP-transduced controls in WR and SHR showed no significant change in HF of pulse interval and LF/HF of pulse interval during the entire observation period.
3.2.4 Respiration
Respiratory rate prior to hKir2.1 and eGFP viral transduction in both rat strains was not significantly different and eGFP expression had no effect on it in either strain (Fig. 3C). Seven days after transduction with hKir2.1 in SHR, but not in WR, respiratory rate decreased significantly and remained depressed throughout the remainder of the observation period (Fig. 3C).
3.2.5 Urine output and water consumption
Within one day after transduction with hKir2.1, water consumption in hKir2.1-transduced WR decreased and returned to the level comparable to eGFP-transduced controls by day 14 (Fig. 5A). In parallel, urine output in WR decreased rapidly and recovered to pre-injection levels by day 14 (Fig. 5C). In SHR there was a clear trend to the opposite effect; for example, water intake in hKir2.1-transduced animals at almost all time points was higher than in eGFP-transduced controls, and their urine output was greater, although these differences were only statistically significant on some days for water intake (Fig. 5B,D).
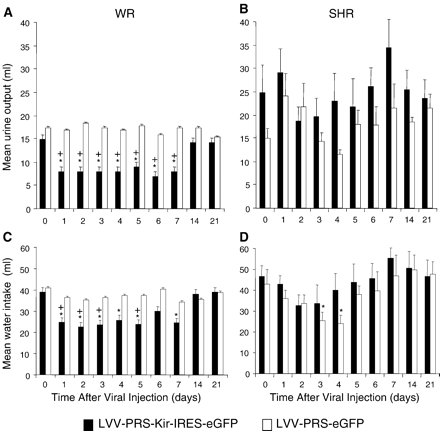
Effects of chronic expression of hKir2.1 on urine output and water intake. Expression of hKir2.1 in WR strain decreased urine output (A) and water intake (C). Changes were significant decrease between 1 to 7 days post-injection and returned to pre-injection levels by day 14. There was no significant change in urine output and water intake (except for days 3 and 4) in the SHR (B, D). eGFP transduced rats of both strains showed no significant changes during the entire observation period.
4 Discussion
The major new finding of this study is that expression of hKir2.1 in A2 neurons leads to a chronic and sustained elevation of arterial pressure in both normotensive WR and SHR with an earlier onset of the effect in SHR. In the SHR this is accompanied by elevations in both VLF and LF but only VLF in WR. In addition, silencing of these neurons increases lability of arterial pressure in both rat strains although there was no change in either HR or cardiac baroreceptor reflex gain. Ventilatory rate was also decreased but this response was confined to the SHR. Finally, silencing of A2 neurons also affected water consumption and urine output in WR but not SHR. Overall, there appear to be multiple and different functional roles of A2 neurons in these two rat strains with distinct underlying neuronal mechanisms.
4.1 Technique considerations
We acknowledge that the inability to measure total gain or maximal gain of the baroreceptor reflex is a disadvantage of the sBRG technique. However, the physiological relevance of the values obtained should not be underestimated because they are around the operating point of this homeostatic reflex. We chose this method as it is non-invasive, allows both chronic and continual tracking of baroreceptor reflex gain giving multiple data points in unrestrained animals and avoids potential confounding influences of vasoactive agents that could exert their own effects at the baroreceptor transducers themselves and/or centrally. We also acknowledge that spectral analysis of blood pressure has its own limitations and additional invasive experiments would be needed to ratify changes in sympathetic discharge by direct recordings.
4.2 Mechanisms by which A2 neurons might regulate arterial pressure in WR and SHR
The arterial pressure increased in both rat strains after expression of hKir2.1 in A2 neurons and we could not reveal any measurable difference in either spread of transgene or basic membrane properties of A2 neurones between the two rat strains. However, our analysis indicates that the autonomic mechanisms underpinning the hypertension and blood pressure lability may differ between WR and SHR. While we found a significant increase in the VLF of SBP in both rat strains after hKir2.1 transduction, only SHR showed elevated LF of SBP. Since these changes occurred in parallel with the rise in arterial pressure (Fig. 2) they might provide clues to the plausible mechanisms of this phenomenon. Indeed, increased lability has been associated with increased VLF of SBP in rats [28,29] and related to changes in vasomotor tone in relation to thermoregulation, neurohumoral agents, sympathetic activity and local metabolic demands [29]. The increase in the LF of SBP in the SHR reflects an increase in sympathetic activity [25] and, interestingly, this effect was absent in the normotensive WR, perhaps indicating the hyperexcitability state of the sympathetic nervous system in the SHR. Previous chemical lesioning studies using 6-OHDA to ablate A2 neurons in Sprague-Dawley rats (normotensive) reported lability in arterial blood pressure but without hypertension [12]. A subsequent study confirmed that A2 lesions evoked chronic lability in arterial pressure for up to 11 months and again without hypertension [14]. Why previous studies [12,14] failed to find hypertension after destroying A2 neurons may relate to the methods adopted. As mentioned in the Introduction section, destruction of NEergic neurons using 6-OHDA is likely to cause plastic changes in nearby neuronal architecture with unpredictable compensatory outcomes. The other factor which could affect the outcome is the impact of 6-OHDA on non-catecholaminergic neurones such as the serotoninergic neuronal processes in NTS [17]. These factors could account for an absence of a chronic change in arterial pressure when neurotoxin 6-OHDA is employed. Interestingly, a transient rise in arterial pressure was evoked 3 h after 6-OHDA treatment but by 24 h arterial pressure was back to control [14] supporting the notion of neuronal compensation.
The increase in blood pressure in our experiments occurred without a measurable change in cardiac baroreflex. The scenario of a change in arterial pressure without alteration in cardiac baroreflex gain is not uncommon as it occurs during physical exercise in both humans and animals [30–32]. We propose that a similar mechanism operates during A2 neuronal silencing in the present study. Therefore, our results suggest that arterial pressure is, in part, determined by the activity of the A2 NEergic neurons in both normo- and hypertensive rats. This notion is consistent with the NTS being a potential site of action of central antihypertensive drugs, such as an α2 agonist clonidine (but recognise also the rostral ventrolateral medulla [33,34]).
It is of interest that intermittent hypoxia, a known stimulant for hypertension and sympathoexcitation, is associated with a reduction in the tyrosine hydroxylase content and its phosphorylation in brainstem catecholaminergic neurons including those in the A2 region [35]. This might be expected to cause a reduction in catecholamine release. In this regard, our attempts to depress the electrical excitability of A2 neurons might, in part, mimic the effect of intermittent hypoxia and hence contribute to the hypertension [35].
4.3 A2 neurons and body fluid homeostasis
Expression of hKir2.1 in the A2 group of WR but, surprisingly, not SHR resulted in a significant decrease in water intake and urine output during the first seven days post-viral transduction. This suggests that A2 neurons have qualitatively different homeostatic functions in these two rat strains. Concerning the reduced urine output in WR, we suggest that this could be a result of vasopressin release. However, whilst we acknowledge that A2 neurons project to the paraventricular nucleus (PVN) [36] and that non-selective NTS cell type lesioning produces a vasopressin-sensitive increase in arterial pressure [37,38], we were unable to detect a change in plasma vasopressin levels (unpublished finding). We speculate that oxytocin might play a role since the A2 projection includes innervation of oxytocin-containing PVN neurons [39] and oxytocin has been associated with hypertension [40] and inhibits drinking [41]. In addition, it is not impossible that silencing of A2 neurons could have an effect on water absorption from the intestine as it has been demonstrated that NTS can modulate intestinal absorption [42].
4.4 Unlikely role of dorsal vagal motor neurons in mediating the hypertension
As mentioned above, the PRSx8 promoter is active in dorsal vagal motor neurons and as their dendrites project into the NTS, some of these cells became transduced with hKir2.1. We do not believe that this had any significant functional implications for the arterial blood pressure effects we report. While we did not assess non-cardiovascular visceral functions that depend on the activity of some vagal motoneurons, such as gastro-intestinal motility, some of these cells are known to project to the heart mediating chronotropic influences. If silencing of these cells due to expression of hKir 2.1 was functionally significant, we should have registered a tachycardia and a corresponding change in HR variability and reduced high frequency in its power spectrum. However, these effects were absent, indicating that the unsolicited transduction of some dorsal vagal motoneurons appeared not to influence the cardiovascular variables described here.
In conclusion, the present study has employed a novel molecular tool to examine the physiological role of A2 neurons. It has demonstrated that these neurons are sympatho-inhibitory in function and play a role in the chronic regulation of the level of arterial pressure in conscious rats. Moreover, silencing of A2 neurons in WR and SHR revealed different autonomic mechanisms for lowering arterial pressure as well as distinct functional roles of these neurons in the two strains of rat. Future studies should seek to determine the factors that control and modulate the activity of A2 neurons, as this appears critical for the chronic maintenance of arterial pressure. As suggested previously[1] factors such as blood flow, arterial oxygen, carbon dioxide or pH may all play vital signalling roles that command the activity of A2 neurons, and hence, arterial pressure.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.cardiores.2007.06.018.
Acknowledgements
Hanad Duale was funded by a MRC PhD studentship. Patrick Howorth is funded by the Wellcome Trust and Anja G. Teschemacher holds a British Heart Foundation Intermediate Fellowship. We are grateful to Dr. Anthony E. Pickering for his assistance in the electrophysiological measurements in PC12 cells. The work was funded by the British Heart Foundation (RG/02/011) and National Institutes of Health (HL033610-18). Julian F.R. Paton was in receipt of a Royal Society-Wolfson Merit Research Award.
References
Author notes
Currently at the Spinal Cord … Brain Injury Research Center (SCoBIRC), University of Kentucky, Lexington, Kentucky, 40536-0509, USA.
AGT and JFRP are equal last authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}