





Abstract
Obesity is strongly associated with the pathogenesis of type 2 diabetes, hypertension, and cardiovascular disease. Levels of the hormone adiponectin are downregulated in obese individuals, and several experimental studies show that adiponectin protects against the development of various obesity-related metabolic and cardiovascular diseases. Adiponectin exhibits favorable effects on atherogenesis, endothelial function, and vascular remodeling by modulation of signaling cascades in cells of the vasculature. More recent findings have shown that adiponectin directly affects signaling in cardiac cells and is beneficial in the setting of pathological cardiac remodeling and acute cardiac injury. Several of these effects of adiponectin have been attributed to the activation of the 5′ AMP-activated protein kinase signaling cascade and other signaling proteins. This review will discuss the epidemiological and experimental studies that have elucidated the role of adiponectin in a variety of cardiovascular diseases.
1. Introduction
Considerable effort has been directed at understanding the mechanisms of obesity in the pathogenesis of cardiovascular disease. This research has led to the concept that adipose tissue is more than just a simple energy storage compartment, but is also an important secretory organ for bioactive molecules referred to as adipokines. Adipokines contribute to the pathophysiology of obesity-linked disorders through their abilities to modulate inflammatory and metabolic processes. Levels of several adipokines (including leptin, tumor necrosis factor-α (TNF-α), plasminogen activator inhibitor type 1, interleukin-1β (IL-1β), IL-6, and IL-8) increase in obesity and tend to function in a pro-inflammatory manner [1,2]. In contrast, levels of adiponectin decrease in obese subjects and this adipokine functions to inhibit inflammatory processes. Clinical and experimental studies suggest that low adiponectin levels contribute to the development of obesity-linked illness including cardiovascular disease, insulin resistance and inflammation. This review focuses on the actions of adiponectin on the cardiovascular system in association with obesity-linked disorders.
Adiponectin, also referred to as ACRP30, AdipoQ and gelatin-binding protein-28 [3–5], is produced in adipocytes and accounts for as much as 0.01% of total plasma protein [6]. The primary protein sequence of adiponectin contains a collagen-like domain at the N terminus and a globular domain at the C terminus, similar to collagens VIII, X and complement factor C1q. The 30 kDa monomers of adiponectin have been shown to aggregate into several polymeric forms in human and mouse plasma, including trimeric, hexameric, and high-molecular weight oligomeric forms [7,8]. In addition to oligomers, adiponectin can also be processed by proteolysis, and a smaller globular domain fragment can be detected in plasma [9]. These various forms of adiponectin are postulated to have distinct signaling effects in the cardiovascular system [7,10,11].
2 Epidemiological studies on adiponectin
The role of adiponectin in obesity, diabetes and cardiovascular disease has been examined through several epidemiological studies discussed below.
2.1 Obesity
Total plasma adiponectin levels typically range from 3–30 μg/ml, in normal human subjects [6,12]. However, levels of adiponectin are significantly reduced in obese subjects compared to non-obese subjects, such that a significant negative correlation is found between body mass index (BMI) and plasma adiponectin levels [6,13]. Adiponectin concentrations have been negatively correlated with percent body fat, waist-to-hip ratio and intra-abdominal fat [13–15]. The reason for this reduction in adiponectin in obese subjects remains unclear but it may be due to either transcriptional suppression or decreased secretion caused by inflammatory cytokines. For example, pro-inflammatory cytokines (such as IL-6) are upregulated in the obese state and cause both a decrease in adiponectin mRNA and a reduction in adiponectin secretion from 3T3-L1 adipocytes [12,16].
2.2 Type 2 diabetes
Studies have addressed the role of adiponectin in type 2 diabetes, a disease that is common to the obese population. Plasma adiponectin levels are lower in patients with type 2 diabetes than in nondiabetic controls among subjects with similar body mass indices [17]. Similarly, individuals with higher adiponectin concentrations appear to be at a lower risk for developing type 2 diabetes [18,19]. In accordance with these findings, other clinical studies indicate that increasing adiponectin is a negative predictor of the development of insulin resistance and type 2 diabetes in BMI-adjusted subjects populations [20–22].
2.3 Cardiovascular disease
The role of adiponectin in the development of cardiac disease remains less clear than it does for metabolic disorders. While some studies indicate that low adiponectin levels are associated with cardiovascular disease, not all studies have been able to show such an association. For example, it has been reported that plasma adiponectin concentrations are lower in patients with clinical manifestations of coronary artery disease than in age- and BMI-adjusted control subjects independent of other risk factors [23–25]. High plasma adiponectin levels are associated both with a lower risk of myocardial infarction in men [26] and a moderately decreased risk for coronary heart disease in male diabetic patients [27]. Adiponectin levels are also reported to rapidly decline following acute myocardial infarction [28]. Collectively, these studies suggest that hypoadiponectinemia is associated with the development of cardiovascular diseases that are prevalent in obese individuals. Consistent with this notion, circulating adiponectin levels are inversely correlated with other cardiovascular risk factors, including hyperlipidemia, high blood pressure and C-reactive protein (CRP) levels [12,29,30]. However, other studies have not been able to draw a link between adiponectin levels and cardiovascular disease status. An association between plasma adiponectin concentrations and the risk of coronary heart disease could not be demonstrated in three recent prospective studies: (i) in the Strong Heart Study, plasma adiponectin levels did not correlate well with the incidence of coronary heart disease [31], (ii) despite a strong correlation of adiponectin levels with adiposity in the British Women's Heart Health Study, adiponectin levels were not predictive of coronary heart disease [32] and (iii) in a large study in British men with coronary heart disease combined with a meta-analysis of previously published prospective studies, the association between adiponectin levels and the risk of coronary heart disease was weak [33]. Therefore, the role of adiponectin in the development of coronary artery disease is controversial, and it appears that low levels of adiponectin will not be a reliable marker for this disease. One explanation for the discrepancy between studies is that heart disease will contribute to impaired renal function and this will confound the analysis due to a reduction in adiponectin clearance by the kidney.
Since hypertrophic cardiomyopathy and pathological cardiac remodeling are also associated with obesity [34,35], the potential role of adiponectin in hypertrophic cardiomyopathy has been analyzed. Low levels of adiponectin are associated with a further progression of left ventricular hypertrophy in patients presenting with hypertension, left ventricular diastolic dysfunction and hypertrophy [36].
Finally, adiponectin levels may influence the development of chronic heart failure, but the epidemiological data are somewhat complex. This is due in part to the fact that while higher body mass indices are a risk factor for heart failure, obesity is a predictor of improved prognoses in patients with established chronic heart failure because wasting is strongly associated with the increased risk of death in the final stages of this disease [37]. In this regard, high adiponectin levels are a predictor of mortality in patients with heart failure [38]. Presumably, this paradoxical relationship exists because high body mass, hence low adiponectin, favors survival in end-stage heart failure. Therefore, future studies should examine adiponectin levels in patients with stable heart failure.
3 Functional studies on adiponectin
A series of experiments in cell culture and animal models have shown that hypoadiponectinemia contributes to a variety of obesity-related diseases including diabetes, macro- and microvascular abnormalities and cardiac pathology (Fig. 1).
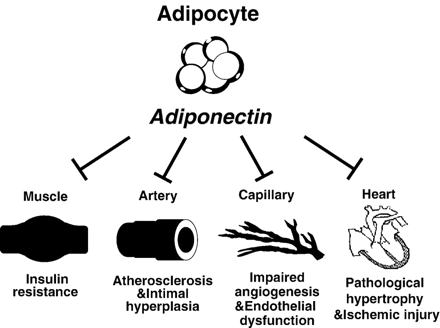
Adiponectin is secreted by adipocytes and has a multiplicity of actions in the cardiovascular system. Adiponectin prevents insulin resistance by enhancing glucose and fatty acid disposal by skeletal muscle. In the heart, adiponectin prevents both pathological hypertrophy and ischemic injury, in part through the activation of AMPK. Adiponectin prevents atherosclerotic progression and intimal hyperplasia by reducing smooth muscle cell proliferation. Similarly, in microvessels and capillaries, adiponectin improves angiogenesis and endothelial function through actions on eNOS and blood vessel growth pathways.
3.1 Glucose metabolism
Cardiovascular disease and insulin resistance are associated with several alterations in metabolism, including changes in the utilization of glucose. Insulin resistant tissues rely more on alternate sources of fuel, since glucose uptake is limited by an impairment of insulin action. High plasma glucose levels result from this decreased glucose disposal and often precede the development of Type II diabetes. Adiponectin has been shown to lower plasma glucose levels in mice, independent of changes in insulin levels [39], suggesting that adiponectin is important for insulin sensitivity. In addition, administration of adiponectin increases fatty acid oxidation in muscle and leads to a reduction in plasma free fatty acids, triglycerides and glucose [9]. These changes are suggestive of an insulin sensitizing action of adiponectin. Analyses of mice deficient for adiponectin confirm the role of adiponectin in insulin sensitivity. Adiponectin knockout (APN-KO) mice are viable and morphologically normal, however they exhibit diet-induced insulin resistance when fed a high fat/sucrose diet [40]. While these data support the notion that adiponectin functions to protect against the development of insulin resistance and diabetes, other strains of APN-KO mice exhibit different insulin-responsive phenotypes. For example, one line of APN-KO mice is reported to exhibit moderate insulin resistance when fed a normal chow diet [41], while another strain of adiponectin-deficient mice do not display this insulin resistant phenotype [42]. These data suggest that the relationships between adiponectin and insulin sensitivity are influenced by additional genetic loci that are currently unknown.
Adiponectin regulates metabolism and insulin sensitivity, at least in part, by promoting the phosphorylation and activation of AMP-activated protein kinase (AMPK) (a stress-responsive kinase) in skeletal muscle [43,44], liver [44] and adipocytes [45]. AMPK signaling affects many aspects of cellular metabolism including glucose uptake, glucose utilization and fatty acid oxidation. AMPK causes: (1) GLUT4 translocation to the cell surface to accelerate glucose uptake [46,47], (2) phosphorylation of phosphofructokinase-2 to enhance glycolytic disposal of glucose [48], and (3) phosphorylation and inactivation of acetyl CoA carboxylase, which leads to an increase in fatty acid oxidation rates [49,50]. AMPK activation is believed to be mediated by adiponectin binding to the cell surface receptors AdipoR1 and AdipoR2 [51]. Another adiponectin receptor (T-cadherin) has also been identified [52], but its role in activating AMPK and other intracellular signaling pathways remains unclear.
3.2 Atherosclerosis
Increasing evidence from experimental models indicates that adiponectin plays a pivotal role in the development of obesity-related vascular diseases including atherosclerosis. In the early stages of atherosclerosis, circulating monocytes adhere to activated endothelial cells (for a comprehensive review see [53]). Subsequently, monocytes differentiate into macrophages, leading to the accumulation of lipid rich foam cells by an uptake of modified lipoproteins. In vitro experiments show that adiponectin regulates many steps in this atherogenic process (Fig. 2). Adiponectin inhibits nuclear factor-κB (NF-κB) activation, effectively reducing: (i) TNF-α-stimulated monocyte adhesion to endothelial cells, (ii) TNF-α-stimulated expression of adhesion molecules and (iii) expression of the pro-inflammatory cytokine IL-8 in endothelial cells [54,55]. Adiponectin also inhibits the transformation of macrophages to foam cells and suppresses the expression of class A scavenger receptors in human macrophages [56]. Adiponectin reduces TNF-α production in human macrophages [57], and this effect may be mediated by its ability to suppress NF-κB signaling in this cell type [58,59]. Adiponectin also increases the expression of the anti-inflammatory cytokine IL-10 and the tissue inhibitor of metalloproteinase-1 in macrophages [60].
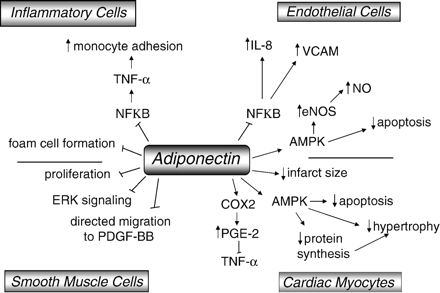
Signaling pathways downstream of adiponectin in cells of the cardiovascular system. Adiponectin has anti-inflammatory effects due to suppression of NF-κB signaling in monocytes/macrophages and also reduces the progression of atherosclerotic lesions through suppression of NF-κB in endothelial cells. In addition, adiponectin signals through the AMPK pathway to reduce endothelial cell apoptosis and to promote nitric oxide production. In the heart, adiponectin activates AMPK and decreases the hypertrophic response through suppression of protein synthesis. COX-2 activation by adiponectin decreases expression of TNFα in the heart. Finally, adiponectin acts in smooth muscle cells to prevent atherosclerotic proliferation and migration of smooth muscle cells.
In vivo experiments have shown that adenovirus-mediated adiponectin overexpression reduces the formation of atherosclerotic lesions in the aortic sinus of the apolipoprotein E knockout (ApoE-KO) mouse (a model of atherosclerosis) [61]. This anti-atherogenic action of adiponectin is accompanied by reductions in the expression of class A scavenger receptors, TNF-α and vascular cell adhesion molecule-1. In agreement with this finding, ApoE-KO mice that overexpress an adiponectin transgene are protected against the development of atherosclerosis compared to untreated ApoE-KO mice [62].
3.3 Angiogenesis and endothelial function
In addition to the actions of adiponectin on atherosclerosis in the vasculature, it has also been shown that adiponectin has effects on angiogenesis and endothelial function. Obesity and diabetes are associated with endothelial dysfunction, microvascular rarefaction and reduced collateralization [63–68], suggesting vascular abnormalities occur in the pathogenesis of these diseases. A series of in vitro and in vivo studies suggest that adiponectin has protective actions on endothelial cells, and may therefore protect against the pathogenic effects of obesity on vascular function. Similar to the effects of adiponectin in muscle, it has been suggested that adiponectin acts through the AMPK signaling pathway in the vasculature (Fig. 2).
AMPK has been identified as a regulator of endothelial cell nitric oxide synthase (eNOS) activation as well as a number of cellular responses that are important for angiogenesis [69–75]. More recently, it has been recognized that adiponectin-mediated activation of AMPK is important for endothelial function and angiogenesis. It has been shown that adiponectin stimulates nitric oxide production in endothelial cells through an AMPK-dependent phosphorylation and activation of eNOS [70,76]. Globular adiponectin increases eNOS expression/activity in endothelial cells and also improves OxLDL-induced suppression of eNOS activity [77,78]. These studies suggest that adiponectin may play a critical role in maintaining endothelial function and vascular tone. Further evidence supports this role for adiponectin since the APN-KO mice exhibit an impaired endothelial-dependent vasodilation on an atherogenic diet [79]. Adiponectin also has anti-apoptotic actions in endothelial cells that are dependent on the induction of AMPK signaling [80], and globular adiponectin is reported to inhibit angiotensin II-induced apoptosis in human endothelial cells [81].
In addition to the anti-apoptotic actions of adiponectin in the vasculature, evidence suggests that adiponectin promotes growth of new blood vessels. Adiponectin stimulates endothelial cell migration and differentiation into capillary-like structures in vitro through activation of AMPK signaling [70]. Adiponectin supplementation has also been shown to stimulate blood vessel growth in both mouse Matrigel plug implantation and rabbit corneal models of angiogenesis [70]. APN-KO mice display impaired recovery of hindlimb ischemia as evaluated by laser Doppler flow method and capillary density analyses, whereas adenovirus-mediated supplement of adiponectin accelerates angiogenic repair in both APN-KO and WT mice by promoting AMPK signaling [82].
3.4 Vascular remodeling
Adiponectin may also modulate smooth muscle cell (SMC) growth in the development and progression of vascular lesions (Fig. 2). In vitro studies have shown that adiponectin can suppress the proliferation of SMCs and inhibit their directed migration to platelet-derived growth factor-BB [83]. This study also showed that adiponectin inhibits growth factor-stimulated extracellular signal-regulated kinase (ERK) signaling in human aortic SMC. In another study, adiponectin was found to inhibit SMC proliferation through its ability to bind various growth factors and interfere with their ability to activate receptor-mediated cellular responses [84]. Therefore, adiponectin may impair growth signaling pathways in smooth muscle cells and prevent the proliferation of SMCs associated with vascular lesions. The results of in vivo studies are consistent with this proposed inhibition of SMC growth by adiponectin. For example, APN-KO mice exhibit increased neointimal hyperplasia and proliferation of SMCs following acute vascular injury [41,85]. Conversely, adenovirus-mediated adiponectin expression reduces the increase in neointimal thickening observed in APN-KO mice [85].
3.5 Hypertrophic cardiomyopathy
Recent experimental studies have shown that adiponectin influences cardiac remodeling and functions to suppress pathological cardiac growth. In response to pressure overload caused by aortic constriction, APN-KO mice have enhanced concentric cardiac hypertrophy and increased mortality [86,87]. Adenovirus-mediated delivery of adiponectin has been shown to attenuate this cardiac hypertrophic in response to pressure overload in APN-KO, wild-type and diabetic db/db mice [86]. APN-KO mice also exhibit increased cardiac hypertrophy in response to angiotensin II infusion, while adiponectin overexpression reduces the hypertrophy in this model [86].
Adiponectin's actions in the setting of cardiac hypertrophy can be attributed to the modulation of intracellular growth signals in cardiac cells, including the AMPK signaling cascade (Fig. 2). While AMPK activity increases through phosphorylation as the heart undergoes a pressure overload hypertrophy [88], this increase in AMPK phosphorylation is attenuated in APN-KO mice [86]. Experiments in rat neonatal cardiac myocytes show that adiponectin activates AMPK, and inhibits the hypertrophic response to α-adrenergic receptor stimulation [86]. The inhibition of hypertrophic growth by adiponectin can be reversed by transduction with dominant-negative AMPK, providing further evidence that adiponectin acts through the AMPK signaling cascade. Activation of AMPK has been shown to inhibit protein synthesis in cardiac myocytes, which is mediated by a decrease in p70S6 kinase phosphorylation and an increase in phosphorylation of eukaryotic elongation factor-2 [89]. These anti-hypertrophic actions of adiponectin on AMPK are thought to occur via the AdipoR1 and R2 receptors [90]. A number of studies have reported that both of these adiponectin receptors are expressed by cardiac myocytes and heart tissue [51,91–93].
3.6 Myocardial ischemia–reperfusion injury
Obesity-related disorders have a major impact on the incidence and severity of ischemic heart disease [94,95] and evidence suggests that adiponectin is cardioprotective in this setting. Adiponectin inhibits apoptosis in cardiac myocytes and fibroblasts that are exposed to hypoxia-reoxygenation stress [96]. Transduction with dominant-negative AMPK blocks the pro-survival actions of adiponectin, indicating that adiponectin inhibits cardiac cell apoptosis through AMPK-dependent signaling. Similarly, recent work from our group demonstrates that following ischemia–reperfusion, APN-KO mice develop larger infarcts than wild-type mice [96]. These larger infarcts were associated with increased myocardial cell apoptosis and TNF-α expression in the APN-KO mice. Adenovirus-mediated delivery of adiponectin diminished infarct size, myocardial apoptosis and TNF-α production in both APN-KO and wild-type mice. Of note, this study showed that the one-time administration of recombinant adiponectin protein, injected either 30 min before the induction of ischemia, during ischemia or 15 min after reperfusion, resulted in a reduction in infarct size. Thus, short-term administration of adiponectin may have practical clinical utility in the treatment of acute myocardial infarction through the activation of AMPK.
While many studies suggest that cardiac AMPK activation is protective, it is important to note that enhanced AMPK activity in the heart has been suggested to reduce recovery following ischemia/reperfusion injury of ex vivo working hearts. These studies show that AMPK activity and fatty acid oxidation rates rapidly increase during ischemia, which may lead to intracellular acidosis and cell death [49,50,97]. The metabolic role of adiponectin in the ex vivo working heart has not been clearly established, although globular adiponectin has been shown to accelerate fatty acid oxidation independent of AMPK signaling [98]. Therefore, further experiments are required to elucidate the mechanism of adiponectin in the setting of ischemia/reperfusion injury and the putative role of AMPK in cardiac recovery.
The protective action of adiponectin against myocardial ischemia–reperfusion injury also appears to be mediated by its ability to activate cyclooxygenase-2 (COX-2) in cardiac cells [96]. COX-2 and its metabolites have been shown to be required for late preconditioning and play important protective roles in myocardial ischemia–reperfusion damage [99–103]. Recent clinical trials reveal that treatment with selective COX-2 inhibitors resulted in an increased risk for cardiovascular complications [104,105], further implicating a role of COX-2 in protection against ischemia induced injury. The upregulation of COX-2 by adiponectin lead to an increase in prostaglandin E2 (PGE2) synthesis and an inhibition of lipopolysaccharide (LPS)-induced TNF-α production [96]. These findings in cardiac myocytes and fibroblasts are consistent with findings in monocytic cells [96,106,107]. Pharmacological inhibitors of the COX-2-PGE2 pathway were found to reverse the inhibitory effects of adiponectin on LPS-induced TNF-α production in cardiac cells [96]. Of note, COX-2 inhibition had no effects on adiponectin-mediated AMPK activation or inhibition of apoptosis in cultured cardiac cells [96]. As well, AMPK-inhibition had no effect on COX-2 induction by adiponectin or on the suppressive effect of adiponectin on TNF-α production caused by LPS [96]. These findings suggest that adiponectin protects the ischemic heart from injury through the activation of independent pathways involving both AMPK-mediated anti-apoptotic actions and COX-2-mediated anti-inflammatory actions (Fig. 2).
3.7 Viral myocarditis
Finally, the role of adiponectin in viral myocarditis has been explored using a mouse model [47]. Leptin-deficient, ob/ob mice inoculated with encephalomyocarditis virus develop severe myocarditis, which is associated with an increased number of apoptotic and infiltrating cells. This phenotype can be minimized by daily subcutaneous injection of adiponectin over an 8-day period, suggesting that treatment with adiponectin can inhibit the development of acute myocarditis.
4 Conclusion
Adiponectin is an adipose tissue-derived hormone that exhibits diverse protective properties on the heart and blood vessels (Fig. 1). Adiponectin may contribute to the regulation of vascular homeostasis by its ability to affect several signaling pathways in the vessel walls and modulate excess inflammatory responses (Fig. 2). In the heart, adiponectin serves as a regulator of cardiac injury through modulation of pro-survival reactions, cardiac energy metabolism and inhibition of hypertrophic remodeling. Many effects of adiponectin in the cardiovascular system correlate with the activation of both AMPK and COX-2. Further evaluation of the biologically active forms of adiponectin and the adiponectin receptor-mediated signaling events in cardiovascular tissues should lead to a better understanding of how obesity affects the cardiovascular system.
Acknowledgements
This work was supported by NIH grants HL86785, HL77774, HL81587 and AG15052 to K. Walsh. N. Ouchi was supported by a Department of Medicine Pilot Project Grant from Boston University. Post-doctoral fellowship support for T.A. Hopkins is provided by the Canadian Diabetes Association and the Alberta Heritage Foundation for Medical Research. R. Shibata was supported by grants from the American Heart Association Postdoctoral Fellowship Award, Northeast Affiliate. N. Ouchi is supported by an American Heart Association Scientist Development Grant, Northeast Affiliate.
References
Sattar N., Wannamethee G., Sarwar N., Tchemova J., Cherry L., Wallace A.M. et al. Adiponectin and coronary heart disease: a prospective study and meta-analysis. Circulation in press.
Author notes
Time for primary review 22 days

{kind=link}
{kind=link}