





Abstract
Activation of the immune system and derangement of cardiorespiratory neural control are established elements of the complex pathophysiology of chronic heart failure (CHF). The magnitude of these abnormalities relates to disease progression and mortality. Less clear is the origin of these derangements and the sequence of triggering mechanisms in the course of the natural history of CHF. To date, immune activation and autonomic imbalance have been considered independently; we hypothesise they are closely related. Damaged heart muscle through autonomic afferents triggers functional and structural changes in the central nervous system, in part related to inflammatory processes. The altered function of the autonomic centres is expressed as a reduction of central parasympathetic tone. Diminished cholinergic signalling (mainly nicotinergic) activates inflammation and stimulates immune response. These two phenomena predict prognosis and represent therapeutic targets in the syndrome of CHF.
1. Introduction
The clinical syndrome of chronic heart failure (CHF) has traditionally been linked to malfunction of the heart as a pump, usually caused by dysfunction of the ventricular myocardium. There is now mounting evidence to show that the complex pathophysiology of CHF begins with an abnormality of the heart, but involves dysfunction of most body organs, including the cardiovascular, musculoskeletal, renal, neuroendocrine, haemostatic, immune, and inflammatory systems [1,2].
Three of these systems are of particular significance, as they constitute universal mechanisms that relate to survival of the species; these are conserved in evolution. They are: i) the neurohormonal system maintaining the blood pressure and peripheral perfusion in the optimal range [3], ii) the immune system protecting the body from the intrusion of external microorganisms [4,5], and iii) haemostatic mechanisms limiting excessive bleeding [6–8]. The preservation of arterial pressure is a fundamental necessity for the maintenance of the circulation, and during evolution, specific mechanisms have developed to achieve this aim even in unfavourable environmental conditions [3]. The key mechanism is the functioning of the autonomic nervous system which directly affects the heart and blood vessels, and indirectly modulates the blood volume by interacting with the renin–angiotensin–aldosterone (RAA) system and vasopressin signalling [3].
CHF is characterised by a dysregulation of the immune response, and autonomic sympathetic/parasympathetic imbalance accompanied by concomitant abnormalities of reflex cardiorespiratory control [2,5,9–11]. The degree of immune activation and autonomic imbalance is related to CHF severity, disease progression and death [5,9,12–14]. The origins of these derangements and the sequence of their occurrence in the course of natural history of CHF remain largely unknown. Whether they are merely epiphenomena related to CHF severity, or constitute critical pathological mechanisms leading to CHF deterioration is not established.
Although in the context of CHF, immune activation and autonomic imbalance have in general been considered separately, there is recent evidence from experimental studies and non-cardiological clinical settings suggesting that these two phenomena might be closely related [15–18]. In clinical studies of heart failure, immune activation and autonomic changes occur concurrently [14,19–21]. High levels of circulating tumour necrosis factor (TNF)-α correlate with increased serum levels of noradrenalin in patients with both mild-to-moderate [19,20] and severe CHF [22]. In patients with decompensated heart failure, increased interleukin (IL)-6 levels are related to deranged autonomic control as assessed by impaired heart rate variability [21].
Our hypothesis (Fig. 1), put simply, is that myocardial damage, due to either ischaemia or other pathological processes, elicits a reflex neural response, with signals, triggered locally in the injured heart, and being transmitted through autonomic afferents to the brain. It results in functional and structural changes within the central nervous system (CNS), in part related to inflammatory processes [16]. These modify the function of autonomic centers, resulting in centrally depleted parasympathetic tone [17]. Reduced cholinergic (mainly nicotinergic) signalling activates inflammation and stimulates immune response [15,18].
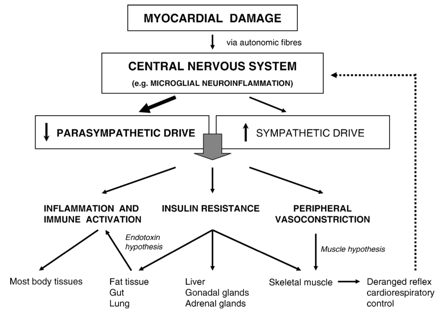
Depiction of hypothetical mechanisms linking centrally depleted parasympathetic drive due to myocardial damage with the progression of chronic heart failure.
2 Inflammatory immune activation in chronic heart failure
A feature of CHF is immune activation [5,11], with pro-inflammatory cytokines (e.g. TNF-α, IL-1, IL-6) overexpressed both in the systemic circulation [23] and locally in the failing myocardium [24]. Sustained overexpression of inflammatory mediators contributes to the development of central and peripheral manifestations of the syndrome of CHF [5,11,23–25] (for extended review see [5,11,25–27]).
Pro-inflammatory cytokines unfavourably affect left ventricular function, exert negative inotropic effect [28], induce abnormalities in cardiac metabolism and energetics, and promote myocardial remodelling [5,11,29,30]. The result is cardiomyocyte hypertrophy [31], necrosis and apoptosis [32,33], and changes in the extracellular myocardial matrix [34]. Additionally, activation of the immune response promotes the development of endothelial dysfunction, general body wasting, skeletal muscle apoptosis, and anorexia in CHF [23,24,35,36]. Many patients with advanced heart failure develop cardiac cachexia, where circulating cytokine levels are the highest [36,37], and cachexia is associated with a particularly poor prognosis [38]. Administration of IL-6 over 7 days, in doses such that circulating IL-6 levels resemble those seen in CHF, results in myocardial failure accompanied by respiratory and peripheral skeletal muscle atrophy [39]. Elevated levels of pro-inflammatory cytokines and their receptors (IL-6, TNF-α, soluble TNF receptors 1 and 2) are strong predictors of increased mortality in patients with CHF, independently of conventional prognostic markers [12,40–43].
Interestingly, pro-inflammatory cytokines exert a variety of biological actions, some of which may appear to be protective for the cardiovascular system [44,45]. The activation of the IL-6-gp130 signalling pathway inhibits doxorubicin-induced cardiomyocyte apoptosis, inactivates caspase 3 [46], and prevents the progression from heart hypertrophy to heart failure at an early stage of pressure overload [47]. It is suggested that the IL-6-gp 130 signalling pathway might be involved into the maintenance of myocardial homeostasis, being an element of complex mechanisms enabling to switch between cardiac hypertrophy, cytoprotection and cellular repair [27,48]. In some circumstances, also TNF-α confers cardioprotective effects, in some of which downstream signalling with NF-κB is involved [44,49]. Mice genetically deficient for the TNF receptors type 1 and 2 demonstrate greater infarct zone and exaggerated apoptosis due to myocardial ischaemia as compared to wild-type animals [50]. In turn, mice genetically deficient for TNF-α demonstrate higher mortality during viral myocarditis as compared to wild-type animals, and an exogenous TNF-α improves survival in a dose-dependent manner [51].
3 Mechanisms of immune activation in chronic heart failure
The origin of immune activation is still uncertain. There are at least five hypotheses. The first is that the failing myocardium itself is the principal source of cytokine production [24]. Pro-inflammatory cytokines produced within the myocardium due to ischaemia or mechanical stress have been shown to contribute to unfavourable remodelling in failing heart [27,52,53]. However, the myocardial production of cytokines is rather a localised phenomenon, as studies have failed to demonstrate the spillover of TNF-α from the heart [54,55]. The vast majority of pro-inflammatory mediators present in the systemic circulation are presumed to be secreted by circulating immune cells. Direct stimuli triggering their activation are unknown. The second hypothesis (the endotoxin hypothesis) proposes that circulatory decompensation results in augmented intestinal translocation of bacterial endotoxin (lipopolysaccharide, LPS) into the systemic circulation, which activates circulating immune cells [4,56]. Patients with decompensated CHF have elevated levels of endotoxin and pro-inflammatory cytokines, which normalise following diuretic therapy [56]. LPS levels in the hepatic vein are higher than in the left ventricle of CHF patients, implicating that gut/liver may be a potential source of circulating LPS [55]. In the study of Aker et al., rabbits with heart failure induced by left ventricular pacing demonstrated increased levels of TNF-α both in serum and hepatocytes, and elevated intestine endotoxin levels, as compared to sham-operated animals [57]. There were no differences in the amount of TNF-α protein in heart tissue between rabbits with or without CHF [57]. These data support the theory that the origin of circulating pro-inflammatory mediators in the course of CHF is peripheral.
The third hypothesis assumes that the primary source of pro-inflammatory mediators is body tissues which are exposed to hypoxia [58,59]. The fourth notion is that immune activation seen in CHF is a consequence of the long-term neurohormonal overactivation and exaggerated stimulation of the sympathetic system [60]. This mechanism may be analogous to that demonstrated in exercise models, where a prolonged and strenuous effort, accompanied by increase in sympathetic tone, is followed by an inflammatory immune response [61–63]. There is enhanced expression of pro-inflammatory mediators both in peripheral blood and skeletal muscles [61–63]. The fifth and novel hypothesis, put forward in this article, is that the initial mechanism triggering inflammatory processes in heart failure is secondary to the central suppression of parasympathetic nervous system.
4 Autonomic sympathetic/parasympathetic imbalance in chronic heart failure
Chronic autonomic sympathetic/parasympathetic imbalance constitutes a fundamental element of CHF pathophysiology [1,2,9,13,64], and is accompanied by abnormalities of reflex cardiorespiratory control (i.e. impaired baroreflex, overactivated ergoreflexes, increased peripheral and central chemosensitivity) [10,65,66].
Autonomic imbalance, in favour of sympathetic tone which is accompanied by depleted vagal drive, occurs at an early stage in the natural history of CHF, and precedes other major derangements, including immune and hormonal pathologies. In an experimental canine model of tachycardia-induced non-ischaemic CHF, parasympathetic tone (expressed as high-frequency component of spectral analysis of heart rate variability) decreased on the third day after induction of cardiac dysfunction [67], and preceded sympathetic activation [68]. In patients with asymptomatic left ventricular dysfunction, neurohumoral activation (as evidenced by increased levels of norepinephrine) precedes the development of symptoms and is related to poor survival [69–71].
Analogously, increased sympathetic tone and depleted parasympathetic drive are present at the early stage of symptomatic CHF, when left ventricular function is only mildly impaired [72,73]. Changes in autonomic balance are seen irrespectively of CHF aetiology [72–74]. Moreover, in apparently healthy subjects, the heart rate profile during exercise and recovery, reflecting depleted parasympathetic tone (i.e. an increased resting heart rate, an insufficient chronotropic response to exercise, an insufficient reduction of heart rate after cessation of exercise), is strongly related to increased mortality due to sudden death, which itself is frequently the first manifestation of cardiovascular pathology [75].
The clinical and prognostic significance of sympathetic overactivation in CHF has been clearly established [9,13,76]. β-adrenergic blockade has become an element of standard CHF therapy [77–79]. In contrast, the reduction in parasympathetic tone in CHF, although demonstrated more that 30 years ago [80], has received less attention. Reduced parasympathetic tone (i.e. blunted baroreflex gain, impaired indices of heart rate variability) predict poor outcome in patients with CHF and in subjects after myocardial infarction [81–83]. In a canine experimental model, pharmacological blockade of vagal reflexes with atropine results in a worsening of existing ventricular arrhythmias leading to sudden cardiac death [84], whereas vagal activation due to direct electrical stimulation of efferent vagal fibers prevents ventricular fibrillation and sudden cardiac death during induction of acute myocardial ischaemia [85].
5 Heart damage followed by changes in the central nervous system
Heart dysfunction due to damaged myocardium is the first direct trigger of the body changes in heart failure which define its natural history. The sequence of subsequent events remains enigmatic.
Recent experimental data reveal a significance of changes within the CNS, occurring even in the early stage of the natural history of heart failure, for example directly after acute myocardial infarction [86]. It is mainly due to the presence of autonomic nervous routes (the cardiac branch of the vagal nerve and the cardiac sympathetic nerves) which connect the heart and the brain, and therefore can convey both ascending and descending information between the myocardium and the CNS [87–90]. There is evidence showing that at an early stage of myocardial dysfunction there is transmission of a neural signal in a reflex manner via autonomic fibers from the diseased heart to the CNS [86–88,91].
Both chemical mediators released during ischaemia and mechanical stimuli are able to modulate the electrical activity of cardiac autonomic sympathetic and parasympathetic afferents [87–89,92–94]. Moreover, most cardiac afferent neurons transduce multimodal stimuli, and can simultaneously sense both mechanical and chemical changes [90]. The sensitivity of cardiac afferents is enhanced in animals with either pacing-induced or ischaemia-induced heart failure; this phenomenon can be demonstrated with regard to both sympathetic [92–94] and parasympathetic [95,96] afferents.
Hua et al. showed a direct involvement of afferent autonomic fibres in the activation of the nucleus tractus solitarius due to myocardial ischaemia in an experimental rat model [97]. Both the occlusion of the left anterior descending coronary artery and direct intrapericardial application of substances released during ischaemia (adenosine, bradykinin, prostaglandins, 5-hydroxytryptamine) stimulated afferent cardiac autonomic neurons, resulting in a rapid increase in FOS immunoreactivity in the nucleus tractus solitarius [97].
In several experimental and clinical models, acute transient myocardial ischaemia, myocardial infarction and chemical molecules released during tissue ischaemia can stimulate neuronal autonomic afferent pathways originating in the heart, which are able to transmit the signal directly to the CNS, and secondarily increase sympathetic tone [91,98–101]. These reactions are rapid, as in a sheep model of acute myocardial infarction, a marked increase in cardiac sympathetic nerve activity is recorded as early as 30min after induction of myocardial ischaemia and remains sustained for 7 days (till the end of observation) [98]. In a human model, the earliest time-point measured was 2–4 days after acute myocardial infarction; Graham et al. demonstrated an increased sympathetic tone at that time-point, which remained high for at least 6 months [99,100]. There is also evidence that even transient ischaemia occurring spontaneously [102] or elicited during angioplasty [103,104] can trigger a dynamic sympathetic activation.
Volume loading can activate both vagal and non-vagal afferent autonomic afferent fibers which modulate the neural activation within the paraventricular nucleus in rats [89]. In normal dogs, an increase in filling pressure (acute volume expansion) results in a selective activation of afferent cardiac autonomic fibers and potentates the cardiac sympathetic afferent reflex [92]. These experimental observations indicate that pressure/volume overload and/or increased wall stretching due to even transient impairment of left ventricular function and haemodynamic deterioration (e.g. during hypertensive crisis, inflammatory process) can trigger analogous neural reflex mechanisms with the involvement of the same autonomic afferent pathways as in a model of myocardial ischaemia.
Recent experimental studies have identified some of the earliest neural and immune mechanisms occurring immediately after the onset of myocardial infarction in the CNS [16,86,105]. In rats, within 30min of myocardial infarction due to coronary artery ligation, there is a significant increase in plasma TNF-α, and an increase in TNF-α mRNA in left and right ventricle (within and beyond the infarct zone) and in the CNS (hypothalamus) [16,86]. The peak in TNF-α mRNA expression in heart and hypothalamus occurs 24h after an ischaemic event, whereas the greatest increment in plasma TNF-α is observed 4 weeks after myocardial infarction [16]. In a rat experimental model, the post-infarction induction of inflammation in the CNS is rapid and of a selective nature. The increased TNF-α expression is limited to the hypothalamus and not present in the cortex [16]. This pattern of TNF-α change in the brain suggests the involvement of a neural reflex mechanism, based on chemical and/or mechanical stimuli derived from the injured heart [16].
In rats an interruption of cardiac sympathetic fibers inhibits the post-infarction TNF-α synthesis in the hypothalamus, but not in heart and peripheral blood, which indicates that this sympathetic (but not vagal) afferent route is indispensable for the induction of post-infarction inflammatory reactions within the hypothalamus [86]. In contrast, vagotomy abolishes the post-infarction TNF-α increase within myocardium (both left and right ventricles) and peripheral blood, but not in the hypothalamus, confirming the significance of vagal efferent signalling for the inflammatory processes selectively in heart and circulating blood [86].
Haemodynamic changes accompanying myocardial infarction seem to be of minor significance and do not explain the subsequent inflammatory reactions in different body compartments. Both mean arterial pressure and pulse pressure were similar in rats with or without vagotomy or sympathetic innervation, and the presence/absence of sympathetic heart innervation was the only factor that affected the TNF-α synthesis in the hypothalamus [86]. The presence of circulating pro-inflammatory cytokines has no effect on the induction of neuroinflammation. Regardless of the presence of increased plasma TNF-α levels, an interruption of cardiac sympathetic fibers effectively inhibits the post-infarction TNF-α synthesis in the hypothalamus [86].
The paper by Francis et al. is the only experimental study directly showing that ischaemic heart damage through neural reflex mechanisms with the involvement of autonomic afferents can trigger inflammatory reactions in the CNS [86]. However, there is also evidence that myocardial ischaemia or/and abnormal haemodynamics can, acting analogously via autonomic afferents, affect autonomic structures in the CNS, causing alterations in their metabolism, reactivity and function [87,89,91,92,97,87].
Additionally, similar data from other clinical settings may be relevant in this context. It is postulated that analogous reflex mechanisms based on the activation of autonomic afferents and subsequently central structures in the CNS are crucial at early stages in the pathogenesis of non-cardiovascular pathologies, including ischaemia and/or inflammation within the gastrointestinal system or lung diseases [106–109].
6 Functional and structural changes in autonomic centres in chronic heart failure
Changes in the CNS occurring in the course of CHF have received only little attention. The evidence is frequently of an indirect nature, and comes mainly from experimental studies.
It is known that general and regional patterns of cerebral blood flow reflecting brain activation significantly differ from age-matched subjects without CHF [110]. In rats with heart failure due to myocardial infarction, neurons in the paraventricular nucleus involved in modulation of autonomic balance demonstrate increased metabolic activity and long-term neuronal activation [111–113].
Recent data suggest that an increased activation of local RAA system in the brain is involved in the central derangement of autonomic control in heart failure [113–119]. Myocardial infarction in rats results in an increase in AT1 (angiotensin type 1) receptor and angiotensin converting enzyme (ACE) densities in the paraventricular nucleus [117]. Also rats with high-output non-ischaemic heart failure demonstrate an increased density of AT1 receptors in the forebrain [120]. Angiotensin II applied to the paraventricular nucleus potentiates the cardiac sympathetic afferent reflex in rats with heart failure [116]. Transgenic rats after myocardial infarction with selectively deficient angiotensinogen in the brain have reduced sympathetic drive and reduced left ventricular remodelling [121].
The pharmacological reduction of overactivated RAA system selectively in the brain results in the amelioration of autonomic control of the cardiovascular system. Centrally administered ACE inhibitors, AT1 receptor antagonists and mineralocorticoid receptor antagonists inhibit the neuronal activity within the paraventricular nucleus in rats with heart failure [113]. Centrally administered AT1 receptor antagonists diminish enhanced cardiac sympathetic reflex in rats with both ischaemia-induced [114] and pacing-induced CHF [115,116]. Sympathetic nerve activity is reduced and baroreflex sensitivity is partially restored in rats with ischaemia-induced heart failure after an intracerebral administration of both an ACE inhibitor [118] and mineralocorticoid receptor antagonist [122]. Central administration of antisense oligonucleotides targeted against mRNA of AT1 receptor in rats with ischaemic CHF reduces both the resting sympathetic tone and the sympathetic reflex response to epicardially administered bradykinin [119].
Central modulation of the brain RAA system affects not only the central sympathetic/parasympathetic balance, but interferes with inflammatory processes. Centrally administered mineralocorticoid receptor antagonist reduces circulating levels of TNF-α in rats with ischaemic CHF [123]. In normal rats, deoxycorticosterone acetate, the precursor of aldosterone, stimulates TNF-α levels in peripheral blood and tissue TNF-α content in the hypothalamus, pituitary, and myocardium (but not in other peripheral organs), mimicking at least partially the central origin of inflammatory changes seen in CHF [105]. This effect is counteracted by an intracerebral application of mineralocorticoid receptor antagonist [105].
Evidence that inflammatory processes are present in the CNS and underlie malfunction of autonomic centres in CHF is rather scarce [124,125]. Such data come predominantly from clinical models of neurodegenerative disorders (e.g. Alzheimer's disease) [126–133].
All neurodegenerative pathologies are characterised by a generalised chronic inflammation (similar to that in CHF) and a prolonged overactivation of brain macrophages accompanied by an overexpression of pro-inflammatory mediators in the CNS (chronic microglial neuroinflammation) [128–130,132,134]. The latter constitutes a major pathophysiological mechanism directly related to neurodegenerative changes which are seen also in patients with head trauma, epilepsy, AIDS and normal aging [132,134].
Patients with coronary artery disease (CAD) demonstrate analogous patterns of morphological and immune changes in brain tissue to those with Alzheimer's disease [135]. Brain lesions, e.g. senile plaques which are characteristic for patients with Alzheimer's disease, are frequently found in non-demented patients with CAD, and their prevalence is similar in both groups [136,137]. It has also been reported at autopsy that in age-matched non-demented subjects, the prevalence of senile plaques and the extend of microglial activation was higher in patients with heart disease than those without [135]. Thus, neuroinflammation and accompanying changes in the CNS are not restricted to patients with neurodegenerative disorders, and may well be present in subjects with cardiac pathologies. This, in turn, forms a background to explain chronic central autonomic dysfunction. From experimental models of neurodegenerative disorders, it is known that microglial overactivation is followed predominantly by a reduction of central cholinergic neurons [17,138], as cholinergic neurons are particularly susceptible to pro-inflammatory mediators and autoimmune antibodies originating from overactivated microglia [17,131,138]. Thus, chronic neuroinflammation is presumed to promote the development of reduced central parasympathetic drive in patients with Parkinson's or Alzheimer's disease [139], similar to patients with CHF.
7 Cholinergic anti-inflammatory efferent pathway
The recent description of the cholinergic anti-inflammatory pathway (an evolutionarily conservative neural reflex mechanism regulating the magnitude of immune response in many organisms) [15,18,140] has shed some more light on mechanisms that enable immune and autonomic systems to interact with each other. This concept explains why depleted parasympathetic drive may be followed by exaggerated inflammatory reactions.
The fundamental assumption of this theory is the fact that acetylcholine, apart from its central and peripheral neurotransmitter role, is a major anti-inflammatory mediator [15,140–142], being a crucial molecule in autonomic–immune interactions. Non-neural cholinergic system (including structures involved into acetylcholine metabolism and elements of intracellular parasympathetic signalling pathways) is present in most cells and tissues, including immune cells [15,140,143–146]. This implies that effects of cholinergic signalling are a generalised and universal phenomenon.
Stimulation of parasympathetic efferent fibers is followed by anti-inflammatory reactions (e.g. reduced expression of TNF-α), demonstrated also within myocardial tissue in a canine model of chronic heart failure [147]. In contrast, in most studies vagotomy results in the exacerbation of immune response; and these effects are observed in many experimental models, involving various tissues, e.g. peripheral blood, gastrointestinal tract [15,140,142,146]. Anti-inflammatory properties of cholinergic signalling have been also demonstrated in cultures of human peripheral macrophages [142] and murine microglia [148]. The anti-inflammatory effects of acetylcholine are related mainly to nicotinergic signalling (with the involvement of a α7 subunit of nicotinic receptor) [18,141], as in most experiments immunosuppressive activity of augmented parasympathetic drive is blunted by a selective nicotinergic antagonist [15,141,142,146,148].
The only paper with conflicting evidence in this context is the study of Francis et al. who suggest that vagotomy 1h prior to ventricular ischaemia in the rat has anti-inflammatory effects, e.g. diminishing the post-infarction TNF increase in myocardium [86]. It is difficult to explain this finding taking into consideration the evidence for the immunosuppressive effects of cholinergic signalling described at the cellular level [141,142,148], and the lack of data suggesting pro-inflammatory properties of acetylcholine. A possible explanation is that vagotomy had substantial effects on heart rate and haemodynamics which may have determined the early response to vagotomy. The other explanation might be the distinction between acute (within minutes) and chronic (within days and months) effects of vagotomy in animals [149]. As demonstrated by van Westerloo and van der Poll, in a rat model of endotoxemia, vagotomy increases plasma TNF-α when performed 3 days before LPS administration (an effect which is analogous to that described by Borovikowa et al. [142]). In contrast, vagotomy paradoxically diminishes plasma TNF-α when performed 30min directly before LPS administration (which seems to be due to releasing of acetylcholine by nerve endings during peri-vagotomy mechanical manipulations on vagal nerve) [149].
Additionally, autonomic imbalance may cause indirectly anti-inflammatory effects, interfering with insulin metabolism [150,151], modifying endothelial function [152,153] and affecting oxidative stress [154]. Reduced parasympathetic tone is known to increase insulin resistance (resulting in reduced uptake of glucose and free fatty acids by adipocytes) and promote enzymatic pathways leading to lipolysis [150,151]. Parasympathetic depletion is also related to generalised metabolic disturbances, i.e. peripheral insulin resistance in skeletal muscles and myocardium, and increased circulating free fatty acids (pro-atherogenic and pro-inflammatory factors) [150,151]. There are close links between the autonomic system and endothelial function, thus influencing the regulation of vasomotor tone and blood pressure [152,153]. Depleted parasympathetic tone results in an inhibition of nitric oxide synthesis, endothelial dysfunction, vasoconstriction (demonstrated within both the systemic and coronary beds), and increased oxidative stress; all these mechanisms are known to augment inflammatory reactions [152–154].
Whereas cholinergic stimulation results in anti-inflammatory reactions in peripheral tissues as described above, the effect of adrenergic signalling on the immune system is rather equivocal. In some studies, β-adrenergic stimulation is followed by anti-inflammatory reactions [155]. In contrast, chronic β-adrenergic stimulation with isoproterenol results in an overexpression of pro-inflammatory cytokines in rat heart tissue, promoting the development of dilated cardiomyopathy [156]. In a subanalysis of the Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF), β-blockade with metoprolol had no anti-inflammatory effect in patients with CHF [157]. In experimental studies, metoprolol showed no immunomodulatory properties [158,159]. There is increasing evidence for the anti-inflammatory properties of carvedilol, which can suppress cytokine production, diminish expression of activation markers, and reduce NF-κB activity in human T-cells [160]. Carvedilol attenuates catecholamine-stimulated IL-6 synthesis in rat cardiofibroblasts [161]. In a rodent model of myocarditis, carvedilol (but not metoprolol) attenuates the over-expression of myocardial pro-inflammatory cytokines [158,159], and in humans, carvedilol therapy reduces levels of circulating IL-6 [162]. These data suggest that anti-inflammatory properties are not a generalised feature of all β-blockers, but are rather related to special (selective) properties of particular molecules, which may not be related to anti-adrenergic properties.
8 New therapeutic approaches
A consequence of this hypothesis is that the restoration or even augmentation of parasympathetic tone (pharmacologically, when using scopolamine, pyridostigmine or non-pharmacologically, by electrical stimulation of parasympathetic efferents, or exercise training) could be an alternative therapeutic approach not only for the normalisation of autonomic balance, but also for the inhibition of inflammatory processes contributing to the progression of CHF [163–167].
Anti-inflammatory interventions in heart failure (for example, clinical trials testing anti-TNF therapies in patients with CHF: RENAISSANCE, RECOVER, ATTACH) have been prematurely terminated due to lack of clinical benefit [168,169]. These results are not as discouraging as appear at first sight. There are unsolved issues, such as the use of too high doses and the imprecise entry criteria for such therapy. Patients with documented inflammatory processes and severe metabolic derangements would be likely to benefit the most. Finally, it cannot be excluded that a selective approach targeting only one cytokine (TNF-α) may be just too simple and insufficient to counterbalance detrimental pathomechanisms of CHF [168,169]. Much more optimistic are results of the phase II of the trial testing broad-spectrum immune-modulation therapy enhancing natural anti-inflammatory mechanisms; patients with advanced CHF receiving an active treatment demonstrated a reduction in both mortality and hospitalisation rate as compared to a placebo group [170].
Low doses of scopolamine improve heart rate variability and baroreflex sensitivity in patients after myocardial infarction [164] and patients with CHF [163]. Similar autonomic effects can be obtained from administration of pyridostigmine (a reversible cholinesterase inhibitor) in low doses in patients with CHF [165,166]. The potential anti-inflammatory (and insulin sensitising) effects of cholinergic drugs have not yet been tested.
Significant limitations of treatment with acetylcholinesterase inhibitors may be their side-effects, such as increased sweating, salivary and gastric secretion, increased gastro-intestinal and uterine motility, diarrhoea, nausea, vomiting, or headache. These are observed mainly when high doses (up to 1200mg for pyridostygmine) of these drugs are administered, e.g. during the treatment of myasthenia gravis. When low doses of scopolamine or pyridostigmine were applied in patients with CHF, there were no significant differences in the prevalence of side-effects between those treated with an active drug as compared to a placebo group [163–166].
Novel centrally acting acetylcholinestarase inhibitors (donepezil, rivastigmine, galantamine) administered in patients with Alzheimer's disease [171,172] might also be beneficial in patients with CHF who demonstrate centrally depleted parasympathetic tone. These drugs improve parasympathetic drive and e.g. normalise the regulation of cerebrovascular flow, probably secondarily to the improvement of endothelial function [173,174]. There are no data on either safety or efficiency of this type of drugs in patients with CHF.
Potential mechanisms of the benefit of exercise training on survival and hospitalisation in CHF population [175] may be a potent anti-inflammatory [176] effect and inhibition of peripheral muscle afferents, associated with improved autonomic control of the heart and circulation [177].
In a rat experimental model of post-infarction heart failure, long-term electrical vagal nerve stimulation improved cardiac pumping function (lower left ventricular end-diastolic pressure, higher maximum dp/dt of left ventricular pressure), reduced serum levels of norepinephrine and brain natriuretic peptide, as compared to rats with CHF without vagal stimulation [178]. Increased vagal drive markedly improved the survival rate in rats with CHF. There was a 73% reduction in a relative risk ratio of death during a 140-day follow-up in animals with versus without vagal stimulation [178]. Moreover, experimental data suggest that long-term increased vagal tone can reveal anti-inflammatory effects within myocardial tissue [147]. In a dog model of heart failure, the 3-month application of neuroselective electric vagal stimulation was followed by an improvement in left ventricular function and a reduction of mRNA and protein expression of TNF-α and IL-6 in left ventricular tissue [147].
Indirect evidence suggests that selective nicotinergic stimulation might be another approach to the simultaneous augmentation of parasympathetic tone and the suppression of immune response [179]. In an experimental rat model, nicotine applied transdermally inhibits humoral and cell-mediated immune inflammatory responses, decreases the leukocyte migration to the inflammation site, and reduces the inducible expression of pro-inflammatory cytokines [180,181].
Such experiments do not exclude the possibility that beneficial effects on survival are not simply related to improvement in autonomic balance, but may be a consequence of other properties of parasympathetic signalling.
9 Conclusions
It has recently become evident that dysfunction of autonomic nervous system and deranged immune mechanisms are involved in the pathophysiology of the CHF syndrome. Traditionally, these systems have been considered in the isolation, but we hypothesise they closely interact.
A damage to the heart muscle through autonomic afferents triggers functional and structural changes in the CNS, in part related to inflammatory processes. The altered function of the autonomic centres is expressed as a reduction of central parasympathetic tone. Diminished cholinergic signalling (mainly nicotinergic) activates generalised inflammation and stimulates the immune response. These two fundamental phenomena predict prognosis and may represent therapeutic targets in the syndrome of CHF.
We are aware of the limited evidence, mostly of experimental nature, supporting this hypothesis. A crucial piece of evidence needed to verify our hypothesis would be a prospective description of the sequence of changes within the autonomic and immune systems in human heart failure, starting from the early stages of heart dysfunction (due to myocardial infarction, myocarditis, etc.) followed to the advanced stages of heart failure and death. Functional in vivo studies of the CNS and evidence from autopsy (including immunological assessment) would be useful. Finally, there is a need for further data on the effects of in vivo cholinergic stimulation (with an administration of centrally/peripherally acting acetylcholinesterase inhibitors or a direct stimulation of vagal efferent fibres) on the immune system and other metabolic derangements in patients with CHF.
Acknowledgements
EAJ was supported by Postdoctoral Research Fellowship of the Foundation of Polish Science. PAPW holds the British Heart Foundation Simon Marks Chair of Cardiology.
References
Author notes
Time for primary review 18 days

{kind=link}