





Abstract
The documentation of preferential activation of the cardiac sympathetic outflow in patients with heart failure swept aside an entrenched notion that there was a functional sympathetic denervation of the failing heart and provided a theoretical basis for the clinical evaluation of β-adrenergic blocker therapy in this condition. The demonstration that heightened sympathetic nervous system activity is central to the pathogenesis and progression of congestive heart failure (CHF) has now led to the rational use of β-adrenoceptor blockade in CHF. More recently, it has also emerged that the aging heart exhibits some of the characteristic changes in autonomic control which are seen in CHF. Accordingly, alterations in cardiac sympathetic nerve function are now thought to contribute also to the pathophysiology of the aging heart. Furthermore, there is evidence that in humans, sympathoexcitatory rostral projections of brainstem noradrenergic neurons to the forebrain are important in the sympathetic nervous activation of both heart failure and aging. Given these similarities, in this review we compare and contrast the neurobiology of the sympathetic nervous system in the failing heart and the healthy, aging heart, and consider whether the sympathetic activation accompanying aging may, perhaps, underlie and contribute to the neural pathophysiology of heart failure. Our conclusion is, on balance, that this proposition is not supported by the available evidence.
1. Introduction
Since the sympathetic nervous system has such a central place in homeostasis in general, and in circulatory adaptation in particular, it is paradoxical that for so long little was known about the contribution of disturbed sympathetic nervous functioning to the development of disease. In investigative clinical medicine, tests based on the measurement of reflex cardiovascular responses in heart rate and blood pressure to a battery of stressors were for many years the generally adopted methodology. These allowed the detection of disorders characterised by autonomic nervous system failure, but did not have sufficient sophistication to lay bare the more subtle dysfunction seen in chronic heart failure (CHF) and essential hypertension.
There has been much improvement lately in the precision of the available tests for studying the human sympathetic nervous system, and in the level of understanding of sympathetic nervous pathophysiology in cardiovascular disease resulting from their application. However, the applications of these new methods (norepinephrine radioisotope dilution and clinical microneurography) to clinical medicine remains rather limited, primarily confined to the diagnosis of the underlying syndromes of autonomic failure in patients with postural hypotension and syncope. It is in the application of these new methods to research that the real progress has been made. For example, the documentation of preferential activation of the cardiac sympathetic outflow in patients with CHF swept aside the entrenched notion that there was a functional sympathetic denervation in the failing heart. This review seeks to provide an overview of the evidence and responsible mechanisms for changes in the functional state of the sympathetic nervous system in CHF and in the aging process, and finally, to explore the possibility that age-related changes in sympathetic activity contribute to CHF progression.
2. The neural biology of congestive heart failure
An extensive body of evidence, based on human and animal neurochemical studies indicates that CHF is characterised by heightened sympathetic tone. Furthermore, a strong consensus exists as to the adverse influence of sympathetic overactivity on the progression and outcome of CHF [1–3], and the demonstration that beta-blockade is beneficial in CHF is consistent with this view. This concept is entirely consistent with experimental studies that demonstrate pro-arrhythmic, pro-ischemic, and pro-apoptotic effects of exposure to pathological levels of catecholamines. Despite the relative uniformity of these observations, many issues with regard to the biology of the sympathetic nervous system and pharmacological approaches that are available to manipulate its activity (directly or indirectly) remain unresolved.
Early studies directed towards the characterization of sympathetic tone in CHF relied upon the measurement of the plasma catecholamine concentration in peripheral venous blood. Seminal studies, such as those by Cohn and colleagues [1], identified the presence of high plasma levels of norepinephrine in CHF as being associated with an adverse outcome. In parallel, our group developed a radiotracer based approach for the study of both systemic and regional norepinephrine turnover, as a more sensitive guide to sympathetic nervous activity. These studies showed that while total systemic norepinephrine release by the sympathetic nervous system is increased significantly in CHF, a significant proportion of the larger (in magnitude) increase in plasma levels of norepinephrine in CHF is attributable to a reduction in clearance from plasma [4,5].
Although ‘global’ measures (such as the peripheral plasma level of norepinephrine) in CHF indicate heightened sympathetic tone, our group and others have shown that the pattern of activation of cardiac sympathetic nerves is quite heterogeneous in CHF. In particular, the degree of cardiac sympathetic activation can be shown to be far more marked than that in renal, hepatomesenteric, pulmonary and skeletal muscle beds [6,7]. Indeed, the activation of cardiac sympathetic neurons has been shown to be one of the earliest features of neurohormonal modulation in CHF, and in late CHF the extent of cardiac sympathetic activation correlates with an adverse outcome [2,8]. Consistent with heightened cardiac sympathetic nerve activity, evidence for elevated release of neuropeptide Y from the failing heart has also been documented [5,9]. Although, experimentally in animals, recent steps have been made to record cardiac sympathetic nerve traffic electrophysiologically [10], this technique is not suitable for clinical studies. In human CHF, neurophysiological studies have been restricted largely to the recording of muscle sympathetic nerve activity (MSNA), which reveals a similar degree of increased activity to that reflected by radiotracer studies. The use of MSNA recording in contrast to radiotracer methods allows the much greater temporal resolution required for detailed baroreflex studies [11,12].
Despite the documentation of increased cardiac norepinephrine spillover in CHF, several paradoxical findings have been a cause of controversy in the field. In particular the early finding that the myocardial content of norepinephrine was reduced in subjects with CHF led to the conclusion that the heart was ‘functionally’ denervated in CHF [13]. Moreover, when the myocardium is examined histologically to define the pattern of sympathetic innervation, a typical finding is that of reduced innervation density as identified by fluorescent staining or immunostaining for tyrosine hydroxylase, a key enzyme in the synthetic pathway for norepinephrine [14,15].
To understand the basis for this apparent paradox, it is necessary to consider all of the factors that influence the rate of turnover of norepinephrine in CHF. Specifically, the following discussion will review the current state of knowledge with respect to the regulation of the control of efferent sympathetic tone, presynaptic control of norepinephrine release and reuptake of norepinephrine in the CHF paradigm.
2.1. Regulation of sympathetic efferent nerve activity in heart failure
Classical studies of the reflex control of sympathetic and parasympathetic control of the heart have focused somewhat simplistically upon ‘high pressure’ baroreceptors located in the aortic arch and carotid sinus and ‘low pressure’ cardiopulmonary receptors. While the use of traditional models largely centred around blood pressure modulation of sympathetic tone hold true in general under normal conditions, they do not explain why sympathetic activation is sustained in CHF. Furthermore, the classical models of sympathetic control do not account for the regional pattern sympathetic activation in CHF.
In previous studies we observed a significant positive relationship of pulmonary artery pressure or pulmonary capillary wedge pressure with the rate of norepinephrine spillover from the heart [5]. Consistent with this observation, a similar observation has also been made with respect to the relationship between left ventricular filling pressure and MSNA [16]. Furthermore, interventions that reduce cardiac filling pressures may lead to reduced cardiac adrenergic drive, which is observed during systemic administration of sodium nitroprusside [17]. Moreover, these changes were uncoupled from total systemic sympathetic activity, which was activated in response to a modest reduction in mean arterial pressure. Consistent with our findings, Azevedo and colleagues [18] showed that modulation of filling pressures by the application of lower body negative pressure reduced cardiac adrenergic drive in CHF patients, but this was not observed in subjects without CHF. Other vasodilator therapies known to improve outcome in CHF, such as angiotensin converting enzyme (ACE) inhibitors, have also been shown to reduce cardiac norepinephrine release [19]. However, it has also been shown that ACE inhibitors may influence the rate of release of norepinephrine by modifying the putative facilitatory actions of angiotensin II on sympathetic neurons [20].
Aside from the baroreceptor inputs that contribute to the control of sympathetic outflow in CHF, considerable effort has also been directed at understanding the factors that act within the central nervous system. In particular, the role of suprabulbar subcortical monoaminergic neurons and the role of brainstem angiotensin II have attracted interest because of their capacity to regulate sympathetic outflow in CHF. On the basis of studies showing that forebrain and brainstem monoaminergic nuclei modulate sympathetic tone [21], we assessed the rate of norepinephrine turnover in subcortical regions in CHF and healthy subjects. Our study showed that the rate of norepinephrine turnover in subcortical regions in CHF was significantly higher than that in the cortex, and was significantly higher than that in healthy subjects [22]. Moreover, the rate of subcortical norepinephrine turnover was significantly positively correlated with global sympathetic activity, measured by the total body norepinephrine spillover rate. Recently attention has turned to the role of locally produced angiotensin II in the brain as a regulator of sympathetic efferent activity. For example, studies by Wang et al. [23] showed that the adverse hemodynamic and left ventricular remodeling responses to myocardial infarction in rats deficient in brain angiotensinogen were substantially attenuated, an effect that was most likely due to reduced sympathetic activation. In association, recent data suggests that superoxide may be the key mediator of the angiotensin II mediated central stimulation of sympathetic activity [24]. These observations raise the possibility that one of the favourable effects of modulating the activity of the renin–angiotensin system by drugs such as ACE inhibitors could be mediated centrally [25].
2.2. ‘Local’ mechanisms for altered myocardial catecholamine turnover in heart failure
In association with elevated efferent sympathetic nerve traffic, other factors modify the local concentration of norepinephrine within the failing heart. Although the myocardial catecholamine content is reduced in CHF, radiotracer based studies clearly show an increase in the rate of norepinephrine spillover from the heart [5,6]. In agreement with these observations, more detailed kinetic analyses indicate that the rate of flux of norepinephrine into interstitial fluid is inversely correlated with left ventricular performance [26].
To complement these findings, our group and others showed a significant decline in the rate of uptake of radiolabelled norepinephrine during passage across the failing heart [5,27]. These observations have been supported by clinical imaging studies using the norepinephrine analogue 123I-MIBG. In patients with CHF, the uptake of 123I-MIBG has been shown to be significantly reduced, and the extent of this reduction provides prognostic information [28]. The mechanism responsible for these observations is unclear. Studies in animal models of the expression of the uptake-1 transporter ‘NET’ have failed to reveal a reduction in the level of expression of NET mRNA in the stellate ganglia, despite a substantial reduction in myocardial NET binding sites [29,30]. On the basis of these findings it has been suggested that posttranslational modifications may play a major role in regulating NET activity [30]; however, the precise mechanism is not clear. Taken together, the findings of reduced NET expression and/or activity in association with heightened release provide a mechanism for the original observations of reduced myocardial norepinephrine content in the failing heart.
Other factors may also play a key role in the local autocrine/paracrine regulation of sympathetic nerve function within the myocardium. On the basis of studies demonstrating a reduction in innervation density in the failing heart, we hypothesised that a reduction in the myocardial expression of nerve growth factor (NGF) could play a role in the pathophysiology of CHF. Complementary animal and human studies demonstrated a substantial reduction in the myocardial content and rate of release of NGF in CHF [14]. In this context it has also been shown that NGF modifies the expression of NET in cell culture models [31] and that it also increases the degree of functional coupling (or ‘synaptic strength’) between cardiomyocytes and sympathetic neurons [32].
2.3. Role of adrenoceptors in regulating and responding to elevated sympathetic activity in CHF
Within the myocardium postsynaptic adrenergic receptors play a pivotal role in the transduction of sympathetic activation into the modulation of contractility and heart rate. One of the key changes in the failing heart is the down-regulation of β-adrenoceptors, particularly the β1-adrenoceptor [33], probably as a result of exposure to sustained high levels of norepinephrine. In contrast, the β2-adrenoceptor, located at both sympathetic presynaptic and postsynaptic neuroeffector junctions is less prone to down-regulation and may therefore assume a more prominent role in regulating myocardial function in the failing heart [34]. The clinical importance of such findings becomes even more apparent with the recent discovery of functional polymorphisms of the β1, β2, and α2 adrenoceptors. In particular, it has been demonstrated that polymorphisms of the β1 and β2 adrenoceptors may alter the clinical profile of CHF and the clinical response to carvedilol [35,36]. Additionally, it has been suggested that particular polymorphisms of the α2C adrenoceptor may be accompanied by a greater propensity to CHF [37,38].
Postsynaptic adrenoceptors may also play a role in modulating the release of norepinephrine from sympathetic nerve terminals in the setting of CHF, although this remains controversial. We previously investigated the inhibitory activity of presynaptic α2 adrenoceptors in CHF patients by regionally infusing clonidine into the forearm. In CHF patients the expected inhibitory effects of α2 adrenoceptor stimulation on norepinephrine spillover were markedly blunted, raising the possibility that this mechanism could contribute to the increase in cardiac norepinephrine spillover in CHF [39]. In contrast, the role of presynaptic β-adrenoceptors in modifying neuronal norepinephrine release remains more controversial [40,41].
Taken together, these studies indicate that genetically determined variations in adrenoceptor function may provide additional layers of complexity in determining the basis for sympathetic overactivity in CHF, its influence on the progression of CHF, and the response to therapy.
2.4. Additional factors that regulate cardiac sympathetic activity in CHF
Over the past decade, it has become increasingly apparent that sleep-disordered breathing is a frequent occurrence in patients with CHF [42,43]. We previously found that 27% of patients with moderate to severe CHF had evidence of obstructive sleep apnoea and a further 38% had features of central sleep apnoea. The heightened interest in sleep apnoea in CHF as a potential contributor to CHF progression arises from studies showing the presence of sympathetic nervous system activation in CHF patients, probably as a result of recurrent hypoxemia and hypercapnia arising during periods of sleep apnoea [44]. Indeed, studies of overnight urinary norepinephrine excretion showed that higher levels of sympathetic nervous activity were observed in patients with central sleep apnoea and CHF than in those with normal breathing patterns [45], and this was attenuated by the use of continuous positive airway pressure (CPAP). With respect to the heart, our recent studies showed significantly elevated awake levels of cardiac norepinephrine spillover rates in patients with central sleep apnoea compared to patents with normal nocturnal breathing patterns [46]. However, when these data were assessed in the context of the prevailing levels of CHF severity, the magnitude of elevation of filling pressures emerged as the likely driving stimulus. The identification of sleep apnoea as a major pathophysiological factor has resulted in the increasing application of CPAP as a therapy for CHF. Indeed, CPAP has been shown to result in improved clinical outcome [47]. Consistent with this observation, we found that CPAP acutely reduces cardiac sympathetic efferent tone, possibly due to a reduction in the degree of stretch of myocardial or pulmonary venous stretch receptors [48].
2.5. Neurobiology of heart failure summary
The sympathetic nervous system is activated in patients with cardiac failure, with preferential stimulation of the sympathetic outflow to the heart. The mechanism is not certain, but appears to involve a paradoxical response of the low pressure cardiac baroreceptors to chronically increased pressures, mediated centrally by sympathoexcitatory noradrenergic brainstem neurons projecting rostrally to the hypothalamus. The cardiac sympathetic activation, which contributes directly to cardiac myocyte deterioration, progressive heart failure and lethal tachyarrhythmias, is now a target in heart failure clinical care. In this regard, beta-adrenergic blockade is an established therapy, while continuous positive airway pressure and the pharmacological suppression of sympathetic outflow by imidazoline receptor binding drugs are yet to be proven to be beneficial. More recently, it has also emerged that the aging heart exhibits some of the characteristic changes in autonomic control that are seen in CHF. Given these similarities, we will now review the neurobiology of the sympathetic nervous system in the healthy aging heart, as a prelude to considering whether the sympathetic activation accompanying aging may, perhaps, underlie and contribute to the neural pathophysiology of heart failure.
3. Sympathetic nervous system and adrenal medullary changes with healthy human aging
The effect of aging on the human sympathetic nervous system has been much studied but still remains a subject of considerable dispute. The impetus for continuing interest in this topic has come in part from the recognition that in a range of cardiovascular disorders, including essential hypertension, cardiac failure, and ventricular arrhythmias, for all of which incidence rises with age, sympathetic nervous system pathophysiology may be an important causal component [49]. The possible contribution of age-related sympathetic nervous system activation to the neurobiology of congestive CHF is a particular focus of this review. As a prelude it can be stated that the consensus view is that progressive sympathetic activation occurs with aging [50]. This sympathetic nervous system activation, however, does not uniformly affect all sympathetic outflows. Renal sympathetic nervous tone, for example, appears not to increase with aging [50]. The neural outflow to the adrenal medulla also seems not to be enhanced by aging.
In studies performed in men aged 20–30 years and 60–75 years we used isotope dilution methods [51,52] for testing whether age influences regional sympathetic nervous system activity and the secretion of epinephrine to plasma. Measurements were made both at rest and during the application of laboratory stressors known to stimulate the adrenal medulla and sympathetic nervous system.
Additional testing was done to investigate central nervous system (CNS) control of sympathetic outflow in aging. We did this by measuring CNS norepinephrine turnover and sympathetic activity simultaneously in healthy younger and older healthy volunteers. The estimate of CNS norepinephrine turnover was based on the Fick principle, utilising measurements of venoarterial plasma concentration differences for norepinephrine, 3-methoxy-4-hydroxyphenylglycol (MHPG) and dehydroxyphenylglycol (DHPG) across the brain, with arterial and internal jugular venous sampling, the associated internal jugular blood and plasma flows being measured by thermodilution [53,54]. In several clinical contexts, most notably cardiac failure and essential hypertension, we have previously demonstrated the importance of projections of noradrenergic neurons to the forebrain in generating the peripheral sympathetic nervous stimulation [53,54]. Selective analysis of subcortical norepinephrine turnover is made possible by the lateralisation of cerebral venous drainage, demonstrated with technetium radiotracer SPECT scans, which shows suprabulbar, subcortical venous drainage is typically into the left internal jugular vein, and cortical venous drainage, via the sagittal sinus, is usually into the right internal jugular vein [53,54]. In patients with CHF and essential hypertension, there is a marked and selective increase in brain norepinephrine turnover confined to the suprabulbar, subcortical field of venous drainage [53,54].
3.1. Sympathetic nervous system activation at rest
Mean plasma norepinephrine concentration was 66% higher in the older men, attributable to 22% lower norepinephrine plasma clearance (P<0.05) and 29% higher whole body norepinephrine spillover to plasma (difference not statistically significant). Regional venous sampling disclosed that the sympathetic outflow to all organs was not activated by aging [50,51]. Renal norepinephrine spillover was normal in older men (Fig. 1). Spillover of norepinephrine into the hepatomesenteric circulation was elevated in the elderly, 63 ± 13 (mean, S.D.) ng/min versus 29 ± 9 ng/min (P<0.05). Measurements of sympathetic nerve firing rates with microneurography have previously documented activation also of the sympathetic outflow to the skeletal muscle vasculature during aging [50]. While spillover of norepinephrine from the heart was increased in older men, 21 ± 11 ng/min compared with 11 ± 9 ng/min (P<0.05), diminished norepinephrine reuptake appeared to be a contributing factor (Fig. 2), in addition to increased cardiac sympathetic traffic. The extraction of tritiated norepinephrine from plasma during transit through the heart (which is primarily attributable to neuronal uptake into sympathetic nerves) and the intraneuronal metabolism of the tracer to 3,4-dihydroxyphenylglycol (DHPG) after uptake were reduced, documenting that neuronal norepinephrine uptake was reduced (Fig. 2).
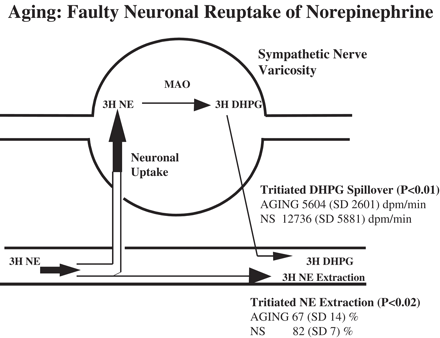
Representation of transcardiac processing of tritiated norepinephrine (3H NE). The majority of tritiated norepinephrine is removed from plasma in transit through the heart via a clearance mechanism involving neuronal uptake by sympathetic nerves. Within sympathetic nerves, 3H NE is metabolised to tritiated 3,4-dihydroxyphenylglycol (3H DHPG) by monoamine oxidase (MAO), with some subsequent release into the venous circulation (coronary sinus). Tritiated norepinephrine uptake by the heart was reduced in older healthy men 67% (S.D. 14%) compared with 82% (S.D. 7%) in young healthy subjects (NS) aged 20–30 years (P<0.02). In parallel, release of tritiated DHPG into the coronary sinus venous drainage of the heart was lower, 5604 dpm/min (S.D. 2601 dpm/min), in older than in younger men, 12736 dpm/min (S.D. 5881 dpm/min)(P<0.01). This provides strong evidence of reduced neuronal norepinephrine reuptake with aging.
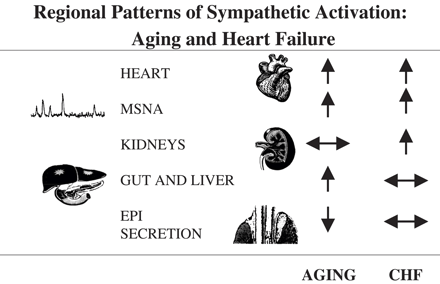
A comparison of the regional patterns of sympathetic nervous activation with healthy aging and in heart failure (CHF). Points of dissimilarity were activation of the renal sympathetic outflow in heart failure but not with aging, activation of the sympathetic nerves to the gut and liver with aging but not in heart failure, and a low rate of adrenal medullary secretion of epinephrine in the elderly, in contrast with heart failure where epinephrine secretion rates are normal or marginally elevated. MSNA: sympathetic nerve firing rates, measured by microneurography, in skeletal muscle vascular efferents; EPI: epinephrine.
3.2. Epinephrine secretion at rest
In contrast to the sympathetic nervous system, for which some outflows become activated during aging, epinephrine release from the adrenal medulla at rest was lower in older men, 112 ± 6 ng/min (mean, S.E.M.) compared with 248 ± 31 ng/min in younger men (P<0.05) [52]. Due to 20% lower epinephrine plasma-clearance in the older men (P<0.01), the reduction in the plasma concentration of epinephrine was proportionally less than that for epinephrine secretion.
3.3. Responses of the sympathetic nervous system to stressors
Measurements were made of epinephrine secretion, whole body and regional norepinephrine spillover rates and haemodynamic responses during mental stress (difficult mental arithmetic), isometric exercise (sustained handgrip) and dynamic exercise (supine cycling). The increase in total norepinephrine spillover to plasma with mental stress was unaffected by age. In contrast, the increase in cardiac norepinephrine spillover was 2- to 3-fold higher in the older subjects (P<0.05) [51].
An almost identical pattern of response was seen with isometric exercise. During cycling, despite total norepinephrine spillover being 16% lower in the older men, cardiac norepinephrine spillover was 53% higher.
Reduced neuronal reuptake of the transmitter [51] appeared to contribute to this augmented sympathetic response in the heart because the transcardiac extraction of plasma radiolabelled norepinephrine was lower in the older subjects during each stressor [51]. Failure of transmitter inactivation at postjunctional receptors with aging due to reduced capture of norepinephrine by neuronal reuptake could amplify an already increased neural signal, and in the presence of myocardial disease could trigger adverse stress-induced cardiovascular events [51].
3.4. Epinephrine secretion with stressors
In the younger men, epinephrine secretion doubled or trebled with mental stress, isometric exercise and dynamic exercise. Epinephrine responses to mental stress and isometric exercise were reduced in older men, being equivalent to only 44% (P<0.05) and 33% (P=0.01) of the corresponding responses in the younger men [52]. The consequences of this diminished adrenal medullary secretory response in the elderly are uncertain, but there may well be a contribution to the subnormal heart rate rise with aerobic exercise, which limits their acute exercise performance, to diminished mobilization of glucose during hypoglycaemia, and to a reduction in endurance exercise capacity associated with deficient adrenergic mobilization of hepatic glucose.
3.5. Influence of aging on CNS control of sympathetic outflow
In this work, sympathetic nervous activity and brain norepinephrine turnover were measured in parallel [53,54]. The overflow of norepinephrine and its lipophilic metabolites MHPG and DHPG into the internal jugular veins, used as a measure of brain norepinephrine turnover, is primarily derived from the noradrenergic neurons of the brain rather than from the sympathetic innervation of the cerebral arterial blood vessels, since ganglionic blockade with trimethaphan does not reduce calculated cerebral norepinephrine turnover, while in patients with pure autonomic failure, who have sympathetic nerve degeneration, transcerebral release of norepinephrine is undiminished [55].
In the study described above we tested whether the sympathetic efferent neuronal activation accompanying human aging originates as a consequence of overactive forebrain noradrenergic mechanisms. We did this by measuring CNS norepinephrine turnover and sympathetic activity simultaneously in healthy younger and older healthy volunteers. Differentiating the pattern of cerebral drainage into the internal jugular veins was of crucial importance in allowing selective measurement of subcortical norepinephrine turnover. Advantage was taken of the fact that the human cerebral venous drainage is asymmetrical to study the regional origins of monoamine spillover into the cerebrovascular circulation [53,54]. Using a technetium-99 cerebral venous sinus scan to delineate the pattern of venous drainage in individual subjects, subcortical and cortical neurotransmitter turnover can be distinguished [53–55]. The commoner pattern is for the right internal jugular vein to have the superior sagittal sinus as its major tributary and the cerebral cortex as its predominant field of drainage; here subcortical venous drainage is into the left internal jugular vein. Sometimes the sinus drainage pattern is reversed, with cortical venous drainage being into the left internal jugular vein. In a minority of cases the drainage pattern is non-lateralizing, with ready admixture of blood occurring at the confluence of the sagittal and straight sinuses.
CNS norepinephrine turnover in the field of an internal jugular drainage was estimated from the combined overflow of norepinephrine and its two major lipophilic metabolites, MHPG and DHPG, into the internal jugular vein. There was a clear cut increase in subcortical norepinephrine turnover in older subjects. The value for norepinephrine turnover in subcortical areas, 317 (S.E. 50) ng/min was three times higher in older than younger men, 107 (S.E. 18) ng/min (P<0.01) [56]. In contrast, values for cortical norepinephrine turnover were similar. A direct, positive correlation existed between subcortical norepinephrine turnover and the level of sympathetic activity in the heart and hepatomesenteric circulation, which was interpreted as suggesting that suprabulbar, sympathoexcitatory projections from noradrenergic neurons of the brainstem to the hypothalamus most probably had an important controlling influence on the sympathetic outflow to these two sympathetic districts; for subcortical norepinephrine turnover versus cardiac norepinephrine spillover; r=0.64, P<0.05 [56–58].
Our results derived here in the setting of aging, and in a range of other contexts indicate the important role played by forebrain noradrenergic mechanisms in the control of human sympathetic outflow in both health and disease. Sympathoexcitatory projections from regions such as the locus coeruleus and A5 region of the brainstem to the hypothalamus and amygdala are no doubt involved, although our methodology does not have the topographic precision to allow study of specific nuclei or their projections. We do acknowledge an inhibitory influence of noradrenergic influences on sympathetic outflow at the brainstem level, within the rostral ventrolateral medullary pressor area (RVLM). These results in humans, however, are in agreement with experimental studies in animals [57,58] that indicate that baroreflex-related bulbar control of sympathetic outflow, although important, can be overridden by forebrain systems, of which rostral projections to the hypothalamus are among the most important.
3.6. Summary position on the question: does age-related sympathetic activation underlie and contribute to the distinctive neurobiology of heart failure?
The prevalence of CHF increases sharply with age. The question has been put: Does the activation of the sympathetic nervous system with aging contribute to the development of CHF, or to its distinctive neurobiology?
Relevant to the second of these questions, Professor Guido Grassi and colleagues from Milan have recently tested whether two conditions other than aging, obesity and high blood pressure, which are known to importantly predispose to the development of CHF and are in both cases characterised by the presence of high sympathetic tone, directly contribute to the level of sympathetic activation present in coexisting CHF [59]. These authors demonstrated that in CHF complicated by obesity, hypertension or obesity-related hypertension there was an additive effect of the sympathetic nervous activation characterising the CHF with that of these other conditions. Among CHF patients, sympathetic nervous activity measured by microneurography was least in lean CHF patients with normal blood pressure (although higher than in healthy people), intermediate in CHF accompanying hypertension or obesity, and highest in CHF accompanying obesity combined with hypertension.
In contrast to these findings, when we tested whether there was an additive influence of age-related sympathetic activation, cumulative with the sympathetic activation of cardiac failure, the answer was negative. The level of sympathetic activation in CHF patients on a cardiac transplant waiting list, these patients being of similar clinical severity (NYHA classes III and IV), was independent of age (Fig. 3). A second line of evidence arguing against age-related sympathetic activation underlying and contributing to the sympathetic activation of cardiac failure was that the organ-specific patterns of activation of the sympathetic nervous system accompanying healthy aging and CHF differed. Two points of divergence were the presence of activation of the renal sympathetic outflow in CHF but not in aging, and the reduced adrenal medullary secretion of epinephrine in aging only (Fig. 1).
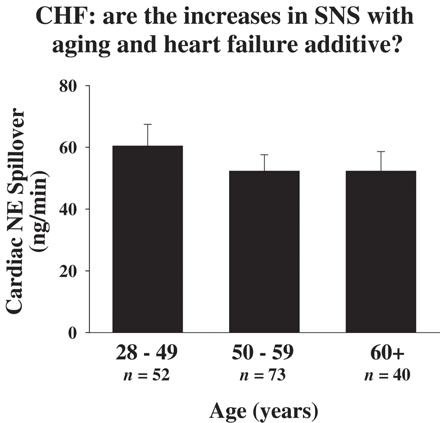
The sympathetic nervous system activation accompanying aging and heart failure was not additive. Cardiac norepinephrine spillover in patients with heart failure was independent of age.
In short, this discord in the neurobiology of CHF and aging, and the absence of an additive effect of aging sympathetic nervous activation in CHF patients, suggests that the upward spiral of CHF prevalence with age derives from causes other than an influence of antecedent, underlying age-related activation of the sympathetic nervous system. Chief among these, no doubt, is the clear and evident impact of aging on coronary artery disease and myocardial infarction incidence rates.
Acknowledgements
Supported by grants from the Atherosclerosis Research Trust, United Kingdom (to DK) and the National Health and Medical Research Council of Australia (to DK and ME).
References
Author notes
Time for primary review 15 days

{kind=link}
{kind=link}
{kind=link}