





Abstract
Objective: Cholesterol-rich membrane domains, which contain the scaffold protein caveolin-1 (Cav-1) (caveolae), represent an important structural element involved in endothelial signal transduction. The present study was designed to investigate the role of these signaling platforms in the generation of endothelial-derived hyperpolarizing factor (EDHF).
Methods: Caveolae were disrupted by cholesterol depletion with methyl-β-cyclodextrin (MβCD 10 mM). MβCD-induced modulation of non-nitric oxide-/non-prostanoid-dependent (EDHF)-mediated vasorelaxation was studied in pig coronary arteries. Effects of MβCD on endothelial Ca2+ signaling and phospholipase A2 (cPLA2) activity were determined using fura-2 imaging and measurement of [3H]-arachidonate mobilization in cultured pig aortic endothelial cells (PAEC). Cellular localization of caveolin-1 and phospholipase A2 was investigated by cell fractionation, and interaction of cPLA2 with caveolin-1 was tested by immunoprecipitation experiments.
Results: MβCD inhibited EDHF-mediated relaxations of pig coronary arteries induced by bradykinin (100 nM) or ionomycin (300 nM) but not relaxations induced by the NO donor DEA/NO (1 μM). Exposure of arteries to cholesterol-saturated MβCD failed to affect EDHF-mediated relaxations. Cholesterol depletion with MβCD did not affect bradykinin or ionomycin-induced Ca2+ signaling in pig aortic endothelial cells, but was associated with enhanced basal and reduced Ca2+-dependent release of arachidonic acid (AA). Cell fractionation experiments indicated targeting of cPLA2 to low density, caveolin-1 rich membranes and immunoprecipitation experiments demonstrated association of phospholipase A2 with the scaffold protein of caveolae, caveolin-1. Cholesterol depletion with MβCD did not disrupt the interaction between cPLA2 and caveolin-1 but prevented targeting of cPLA2 to low density membranes. Exogenous supplementation of arachidonic acid after cholesterol depletion partially restored EHDF responses in pig coronary arteries.
Conclusion: The integrity of caveolin-1-containing membrane microdomains is prerequisite for arachidonic acid recruitment and EDHF signaling in porcine arteries.
This article is referred to in the Editorial by J. D. Paerson (pages 189–191) in this issue.
1 Introduction
Control of vascular tone by endothelial-derived hyperpolarizing factors (EDHFs) is of particular physiological and pathophysiological significance [1–5]. A number of cellular key events involved in the EDHF phenomenon have been identified, but the molecular basis of the non-nitric oxide-, non-prostanoid (EDHF)-mediated relaxation of vascular smooth muscle remains still obscure [2]. Most prominent among the cellular events that have been implicated in EDHF generation and/or action are the activation of potassium channels [6–8] and the formation of epoxyeicosatrienoic acid derivatives (EETs) from arachidonic acid by cytochrome P450 (CYP) epoxygenases [9]. Unequivocally, a key event in the transduction of the primary stimuli eliciting EDHF responses is the elevation of intracellular Ca2+ in the endothelial cells, and mechanical stimuli appear to affect EDHF regulation of vascular tone in addition by promoting the expression of key signaling proteins such as CYP epoxygenases [2]. One key player in endothelial signal transduction is caveolin-1, which is essential for the formation of specific cholesterol-rich microdomains in the plasma membrane (caveolae) [10]. Some of the signaling mechanisms that are of relevance for the EDHF phenomenon such as phospholipase C-dependent Ca2+ signaling [11] and phospholipase A2-arachidonic acid metabolism [12] are likely to depend on caveolae as a signaling platform. Here we tested whether changes in the membrane cholesterol content and disintegration of caveolae structures affect EDHF generation. We obtained evidence for caveolar targeting of endothelial phospholipase A2 and a key role of caveolae in arachidonic acid-dependent EDHF responses of pig coronary arteries. Our data suggest membrane cholesterol and cholesterol-rich membrane microdomains as a key element of EDHF signaling.
2 Experimental procedures
2.1 Measurement of isometric tension in isolated porcine coronary artery rings
Isometric tension of rings derived from porcine right coronary arteries was recorded as described [13]. Coronary rings were stretched in Krebs-Henseleit buffer (in mmol/l: 118.4 NaCl, 5.01 KCl, 1.20 KH2PO4, 2.50 CaCl2, 1.20 MgCl2, 25 NaHCO3, 10.1 glucose) to an initial tension of 10 mN and allowed to equilibrate for approximately 1 h, and then contracted with a thromboxane receptor agonist, U46619 (300 nM), in the presence of l-NNA (250 μM) and indomethacin (10 μM). When the contraction reached a stable plateau, relaxation of the vessels was initiated with either bradykinin (100 nM) or ionomycin (300 nM) to serve as internal control response. Subsequently, rings were repeatedly washed with Krebs Henseleit, and incubated for 1 h with either vehicle plus 10 mM methyl-β-cyclodextrin (MβCD), or 10 mM MβCD-saturated with cholesterol, or vehicle alone. After 1 h, the rings were repeatedly rinsed with buffer, and relaxations in response to bradykinin or ionomycin were again analyzed after precontraction with U46619.
2.2 Preparation of cholesterol–cyclodextrin complexes
Complexes of cholesterol and MβCD were prepared according to Klein et al. [14].
2.3 Cell isolation and culture
Endothelial cells were isolated and cultured from porcine aorta as described previously [15]. Exclusively the first passage was used, and cells were grown to about 80–100% confluency in either 100-mm Petri dishes (6×106 cells/dish) for cell fractionation and immunoblotting experiments, or in 6-well plates (about 106 cells/well) for measurement of arachidonate release.
2.4 Measurement of intracellular calcium in single cells
PAEC were loaded with fura-2-AM (Molecular Probes) as described [16]. Cells were constantly perfused during experiments with buffer (in mmol/l: 137 NaCl, 5,4 KCl, 15 HEPES, and ±2 CaCl2). Excitation light was supplied via a Polychrome II polychromator (TILL Photonics), and emission was detected by a Sensicam CCD-camera (PCO Computer Optics). Fluorescence was recorded at the 340- and 380-nm excitation wavelengths and emission collected at 510 nm. Digital image recordings were evaluated using Axon Imaging Workbench (Axon Instruments). Changes in Ca2+-sensitive fluorescence ratio were used to analyze and compare changes in intracellular Ca2+ (Cai).
2.5 Measurement of arachidonic acid (AA) release
The AA release was determined as described in Ref. [17]. Briefly, PAEC of passage 1 were seeded and grown in 24-well plates overnight. Cells were washed once with PBS (37 °C), incubated with 0.2 μCi [3H]-arachidonic acid in DMEM+10% FCS overnight, and pre-incubated for 1 h at 37 °C with vehicle plus 10 mM MβCD, or vehicle alone (FCS-free DMEM). After pre-incubation, cells were stimulated with U46619 and ionomycin, and the medium was collected at 10 min after stimulation. Samples were centrifuged at 1000×g for 5 min and radioactivity in the supernatant was determined by scintillation counting. Cells were scraped in 0.5 ml 0.1% Triton X-100 for determination the total cellular radioactivity.
2.6 Isolation of caveolin-rich membrane fractions
Caveolae membranes were isolated as described [18] by a method adapted from [19]. PAEC of passage 1 were preincubated with serum-free DMEM in the absence or presence of 10 mM MβCD for 1 h at 37 °C. Subsequently, cells were washed twice with ice-cold PBS, scraped off from the dishes, centrifuged, resuspended in 850 μl of Na2CO3 (0.5 M; pH 11) and homogenized by sonication. The homogenate was mixed with 850 μl of a 90% sucrose solution prepared in MBS (25 mM MES, 0.15 M NaCl; pH 6.5), placed in a AH-650-tube, overlaid by 1.7 ml of 35% and 1 ml of 5% sucrose (each prepared in MBS containing 0.25 M Na2CO3) and centrifuged at 134,000×g for 20 h at 4 °C using the Sorvall AH-650 rotor. Fractions, starting at the top of each gradient, were collected to yield nine samples: 1 ml, from the top, was collected as fraction 1 and subsequently, 0.4 ml fractions were collected as fractions 2 to 9. An aliquot of the samples was used for determination of protein concentration. The proteins in the fractions were precipitated with 25% (v/v) of 50% trichloroacetic acid solution for 25 min on ice. After centrifugation, the precipitated proteins were washed once with 1 ml of ice-cold water, and the pellets dissolved either in 100 μl of Laemmli buffer (heated to 95 °C for 5 min) for immunoblotting or in 100 μl of 1 N NaOH for protein determination. Caveolin-1 was taken as a marker of caveolae, which are typically recovered in low density fractions (#1 and #2). Lactate dehydrogenase, a marker to test for the localization of soluble proteins, was typically retained in fractions 7–9.
2.7 Immunoprecipitation
PAEC were either incubated in serum-free medium in the presence or absence of 10 mM MβCD for 1 h at 37 °C or stimulated with 1 μM bradykinin for 20 min. Afterwards, cells were washed twice with ice-cold PBS, scraped off and centrifuged at 4 °C. Pellets were resuspended in 0.5 ml of ice-cold lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 60 mM OG, and protease inhibitor mixture (complete; Roche Applied Science) and incubated for 15 min on ice. Cell lysates were sonicated and centrifuged to pellet nuclei and unbroken cells. Supernatants were precleared by preincubation with 50 μl of protein A-Sepharose beads for 45 min at 4 °C and incubated with either 10 μl of anti-cPLA2 or with 5 μl of anti-caveolin-1 overnight at 4 °C. Subsequently, 50 μl of protein A-Sepharose beads was added to the immune complex for 2 h at 4 °C. Beads were pelleted, washed three times with ice-cold lysis buffer, resuspended in 50 μl of 2× Laemmli buffer, and boiled at 95 °C for 5 min.
2.8 Immunoblot analysis
Proteins were separated on SDS-PAGE (8% acrylamide for cPLA2, 12% for caveolin-1) and transferred to nitrocellulose membranes (240 mV, 90 min on ice). Nitrocellulose sheets were preincubated for 1 h at room temperature with TBST buffer (in mM: 25 Tris, pH 7.4, 137 NaCl, 2.7 KCl, 0.1% Tween 20) containing 1% BioRad blocking reagent and incubated with primary antibody against either caveolin-1 (1:5000), or cPLA2 (1:100) overnight at 4 °C. After washing with TBST buffer, horseradish peroxidase-conjugated anti-rabbit (1:5,000) against caveolin-1 and anti-mouse (1:1,000) against cPLA2 were used as secondary antibodies. Membranes were detected via Chemi Glow™ West detection system and developed using a Herolab RH-5.2 Darkroom Hood, equipped with an E.A.S.Y 1.3 HC camera (Herolab Laborgeräte, Germany).
2.9 Materials
Tissue culture medium was from Invitrogen; fura-2/pentaacetoxymethyl ester was from Molecular Probes (Leiden, The Netherlands); DEA/NO was from Alexis (Switzerland); anti-Caveolin-1 antibody was from BD Transduction Laboratories (Wien, Austria); monoclonal anti-cPLA2 antibody was from Santa Cruz Biotechnology; Chemi Glow™ West (Biozym, Wien, Austria). All other chemicals were purchased from Sigma (Germany).
2.10 Statistical analysis
Statistical evaluation of the data was performed by using Student's t-test for pair wise comparisons and ANOVA for multiple comparisons. A p value <0.05 was considered statistically significant.
3 Results
3.1 Cholesterol depletion impairs EDHF-mediated responses in pig coronaries
Pig coronary artery rings contracted by the thromboxane mimetic U46619 respond to stimulation of phospholipase C-coupled receptors with a pronounced relaxation. A large part of this relaxation is resistant to suppression of endothelial NO- and prostacyclin production and represents a typical EDHF response. Fig. 1A illustrates this NO/PGI2-independent relaxation of pig coronary induced by stimulation of bradykinin receptors in the presence of l-NNA (250 μM) and indomethacin (10 μM). The experimental protocol was comprised of two test phases, an initial phase to evaluate each vessels response to bradykinin in control conditions (pre-incubation) and a second phase in which the effect of bradykinin was determined subsequently to a 1-h incubation designed to disrupt cholesterol-rich microdomains in the plasma membrane by cholesterol depletion (post-incubation). Aortic rings subjected to this protocol without administration of cholesterol modifying agents displayed a slightly diminished EDHF response in the second test phase (Fig. 1A, upper panel). When aortic rings were exposed to cholesterol-saturated MβCD, EDHF responses were again barely reduced as compared to controls (Fig. 1A, middle panel). By contrast, incubation with the cholesterol scavenger MβCD resulted in a substantial suppression of the EDHF response (Fig. 1A, lower panel). Fig. 1B illustrates a statistical comparison of the effects of cholesterol extraction on EDHF-mediated relaxations as well as on DEA/NO (1 μM)-induced endothelium-independent relaxations. We observed a slight to moderate reduction of EDHF-responses in the post-incubation phases of sham-incubated and MβCD/cholesterol controls. This reductions were statistically significant for MβCD/cholesterol treatment but not for sham incubations. EDHF responses were substantially suppressed after exposure of vessels to cholesterol-free MβCD. Suppression of EDHF responses by MβCD was statistically significant based on a comparison with either sham-incubated or MβCD/cholesterol controls. By contrast, cholesterol extraction with MβCD failed to affect NO-mediated relaxations. Depletion of membrane cholesterol diminished the EDHF-mediated relaxation induced by the Ca2+ ionophore ionomycin (N=18, see below Fig. 6), indicating that cholesterol depletion affects a signaling step downstream of Ca2+ mobilization processes.
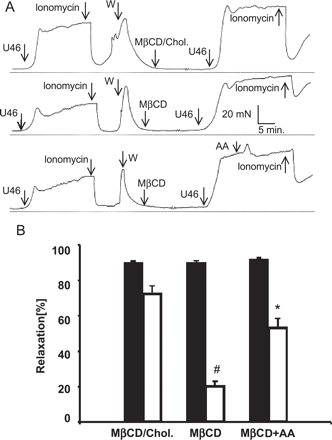
Arachidonic acid supplementation partially restores EDHF responses in cholesterol-depleted coronary arteries. (A) Time courses of a representative set of experiments. Porcine aortic rings were contracted with U46619 (U46, 300 nM), in the presence of l-NNA (250 μM) and indomethacin (10 μM), and relaxation was initiated with ionomycin (300 nM). After a first test phase, rings were repeatedly washed (W) with Krebs Henseleit and incubated for 1 h with either MβCD-saturated with cholesterol (upper panel) or with 10 mM MβCD (lower and middle panel). In one set of experiments, arachidonic acid (AA, 10 μM) was added to the incubation buffer about 15 min prior to the stimulation of rings with ionomycin in the second test phase. (B) Filled and open columns represent EDHF-induced vasorelaxations measured before and after the incubation period (pre- and post incubation), respectively. Mean values±S.E. (N=18). # denotes significant difference versus MβCD/chol (post incubation); * denotes significant difference versus incubation with MβCD (post-incubation).
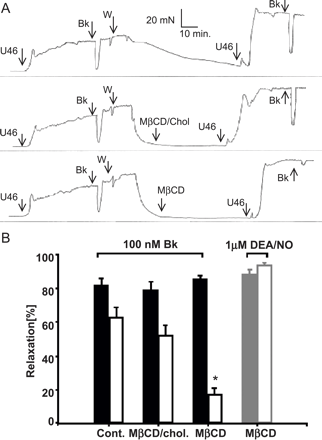
Alteration of membrane cholesterol content impairs EDHF-mediated relaxation. (A) Time courses of a representative set of experiments. Porcine aortic rings were contracted with U46619 (U46, 300 nM), in the presence of l-NNA (250 μM) and indomethacin (10 μM), and relaxation was initiated with bradykinin (Bk, 100 nM). Rings were repeatedly washed (W) with Krebs Henseleit and incubated for 1 h with either vehicle alone (top panel) vehicle plus 10 mM MβCD-saturated with cholesterol (middle panel), or 10 mM MβCD (lower panel). Thereafter, rings were repeatedly rinsed with buffer, and relaxations in response to bradykinin were again analyzed (post-incubation). (B) Bars represent bradykinin (Bk) or DEA/NO-induced EDHF vasorelaxations measured before (pre-incubation; filled columns) and after the incubation period (post-incubation; open columns), respectively. Gray bars represent DEA/NO-mediated relaxation in controls (pre-incubation; filled columns) and in cholesterol-depleted coronary arteries (post-incubation; open columns). Experimental conditions are indicated. Mean values±S.E. (N=18). * denotes statistically significant difference versus controls (post-incubation; Cont. as well as MβCD/chol).
3.2 Cholesterol depletion does not inhibit bradykinin-induced Ca2+ signaling
To test whether cholesterol depletion interferes with cellular Ca2+ signaling processes that are initiated by autacoids, we studied intracellular Ca2+ signals induced by bradykinin in cultured pig aortic endothelial cells (PAEC) using Fura-2 imaging in the classical Ca2+ re-addition protocol. As shown in Fig. 2, MβCD failed to affect bradykinin-induced Ca2+ release from intracellular Ca2+ stores as well as Ca2+ entry into bradykinin-stimulated cells. Interestingly, Ca2+ entry was even slightly augmented in cholesterol-depleted cells. Thus, inhibition of EDHF responses by MβCD appears to be based on interference with a cellular event downstream of autacoid-triggered Ca2+ signaling.
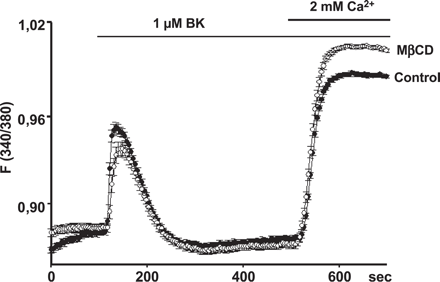
Cholesterol depletion does not impair Bradykinin-induced Ca2+ entry. Time courses of Ca2+-sensitive fura-2 fluorescence ratios (F340/380) recorded in PAEC incubated in FCS-free medium in the absence (control, filled symbols), or presence (open symbols) of 10 mM MβCD for 1 h. Cells were challenged with bradykinin (Bk, 1 μM) in Ca2+-free solution, and extracellular Ca2+ was elevated to 2 mM, as indicated. Mean values±S.E. derived from 23 and 30 cells are shown, respectively.
3.3 Cholesterol depletion affects Ca2+-dependent mobilization of arachidonic acid and disrupts targeting of phospholipase A2 to caveolae
Since arachidonic acid metabolites have been identified as key mediators in EDHF signaling [9,20,21], we hypothesized that MβCD-induced inhibition of EDHF signaling may involve disruption of Ca2+-dependent stimulation of phospholipase A2 and availability of arachidonic acid in endothelial cells. The PLA2 inhibitor AACOCF3 markedly inhibited the NO/PGI2-independent relaxation of pig coronary arteries induced by bradykinin (0.1 mM) from 38±6% to 11±3% (data not shown) demonstrating that EDHF-mediated relaxation of pig coronary is indeed dependent on phospholipase A2 activity.
As illustrated in Fig. 3, cells exposed to MβCD exhibited a significantly higher level of basal arachidonate release, while liberation of arachidonate upon stimulation with the Ca2+ ionophore ionomycin was reduced. Cholesterol saturated MβCD, by contrast, failed to affect arachidonate release (N=6; not shown). Since recent evidence suggested inhibitory coupling of cPLA2 to caveolin-1 (Cav-1) in neuronal cells (23), we tested for targeting of endothelial cPLA2 to caveolae by use of a discontinuous sucrose gradient that enables separation of caveolar membranes [19].
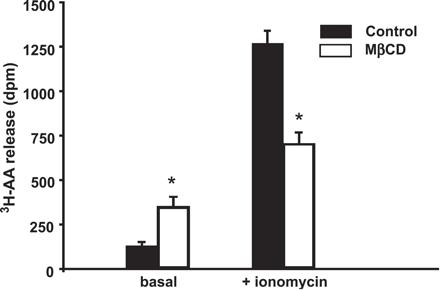
Cholesterol depletion stimulates basal release of arachidonic acid and impairs its mobilization by bradykinin. Arachidonic acid release in non-stimulated endothelial cells (basal, left) and 10 min after stimulation with ionomycin (right, 300 nM). Mean values±S.E. (N=6). * denotes statistically significant differences versus the response obtained under control conditions (filled bars).
Fig. 4A illustrates the distribution of Cav-1, on the density gradient. The scaffold protein of caveolae was highly enriched at the interface between the 5% and 35% sucrose corresponding to the low density membrane fraction #2, designated as fraction enriched in caveolar membranes (CM). Non-raft plasma membrane and intracellular membranes are of higher density and typically found in higher density fractions. Soluble proteins are typically retained in fractions 7–9, as verified with lactate dehydrogenase as marker. A typical feature of caveolae is the dependency of their structure and function on membrane cholesterol. Consistently, cholesterol depletion with MβCD, which is known to disintegrate caveolae [22], resulted in a clear distortion of the cellular localization of caveolin-1. As illustrated in Fig. 4B, a significant amount of cPLA2 was recovered in the caveolin-1 rich fraction (fraction 2; CM) suggesting co-localization of cPLA2 and Cav-1 in caveolar membranes. Extraction of membrane cholesterol with MβCD resulted in a substantial translocation of the protein to higher density fractions. Fig. 4C shows a statistical analysis of the distribution of cPLA2 on density gradients in control and cholesterol depleted cells quantified by densitometric analysis. Cholesterol extraction almost eliminated cPLA2 immunoreactivity in CM membrane fractions, while a clear increase in cPLA2 immunoreactivity was observed in high density fractions 6–8. This result substantiates that cPLA2 is targeted to a cholesterol-dependent, low density membrane structures. Exposure of cells to cholesterol saturated MβCD did not significantly reduce the immunoreactivity of either caveolin-1 or cPLA2 in low density fractions (N=3; not shown). Fig. 4D shows the protein content of the fractions, which was fairly equal throughout fractions 2–9 and was not significantly different between controls and cholesterol extracted cells.
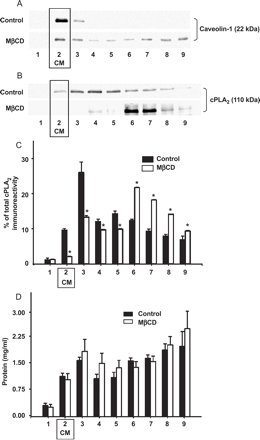
Cholesterol depletion disrupts subcellular localization of caveolin-1 and targeting of cPLA2 to caveolae. Endothelial cells were homogenized and fractionated on a discontinuous sucrose gradient. Nine fractions, with increasing density from 1–9, were collected, and an equal volume of each fraction was separated by SDS-PAGE, followed by immunoblot analysis using antibodies directed against caveolin-1 (A) or cytosolic Ca2+-dependent phospholipase A2 (B). MβCD indicates results obtained with cholesterol extracted cells. Results are representative of five independent experiments. Box highlights the fractions containing caveolin-enriched low density membranes corresponding to caveolae (CM, fraction 2). (C) Statistical analysis of the subcellular distribution of cPLA2 on density gradients. Data represent the intensity of cPLA2 immunoreactivity as determined by densitometry expressed as % of the total immunoreactivity in all fractions of the gradient. Mean values±S.E. (N=5). * denotes significant differences between control and MβCD-treated cells. (D) Distribution of protein concentration in the fractions analyzed for cPLA2 (corresponding to C). Mean values±S.E. (N=5).
To further test for targeting of cPLA2 to caveolae and for a physical interaction between Cav-1 and cPLA2, we performed immunoprecipitation experiments. As shown in Fig. 5, immunoprecipitation with anti-cPLA2 clearly recovered significant caveolin-1 immunoreactivity and, consistently, anti-caveolin-1 co-immunoprecipitated cPLA2. Cholesterol scavenging with MβCD did not disrupt the interaction between cPLA2 and caveolin-1, while bradykinin-induced stimulation of cPLA2 was accompanied by dissociation of cPLA2–Cav-1 complexes as evident from a reduced co-immunoprecipitaion. These data further support the concept that a significant fraction of phospholipase A2 resides in endothelial caveolae and that MβCD interferes with targeting of phospholipase A2 to caveolae, resulting in impaired cPLA2 function and EDHF signaling.

Caveolin-1 and cPLA2 co-immunoprecipitate from pig aortic endothelial cell lysates. Supernatants were immunoprecipitated (IP) with anti-cPLA2 (left panel) or anti-caveolin-1 (right panel), and immunoblotted (WB) to detect caveolin-1 and cPLA2, respectively. Lane 1, proteins from total cell lysates; lane 2–4, proteins precipitated at the indicated experimental conditions; lane 5, background precipitation in the absence of precipitating antibody with protein A-Sepharose beads only. These results are representative of three independent experiments.
3.4 Arachidonic acid supplementation partially restores EDHF responses in cholesterol depleted coronary arteries
To further confirm the above concept, we tested whether exogenous administration of arachidonate as a precursor of EDHF is able to restore EDHF signaling in MβCD-treated cells. Fig. 6A illustrates that ionomycin-induced, EDHF-mediated relaxation of pig coronary arteries was substantially reduced after incubation with MβCD (middle panel) as compared to arteries exposed to cholesterol-saturated MβCD (upper panel). When aortic rings were incubated with arachidonic acid (10 μM) subsequently to depletion of cholesterol with MβCD (lower panel), the ionomycin induced vasorelaxation was significantly enhanced as compared to incubation with MβCD only. A statistical comparison of the effects of cholesterol saturated MβCD, MβCD alone and MβCD plus arachidonic acid on the EDHF responses is shown in Fig. 6B. It is of note that no significant difference was observed between the relaxations (post-incubation) of vessels exposed to cholesterol-saturated MβCD and those observed in cholesterol extracted vessels supplied with exogenous arachidonate. Arachidonic acid (10 μM) neither affected ionomycin-induced relaxation of control vessels nor the direct smooth muscle relaxation induced by DEA/NO (1μM) (N=4; not shown). These data indicate that exogenous arachidonate was able to partially antagonize the inhibitory effect of cholesterol depletion and, thus, point to a role of cholesterol rich membrane domains in EDHF signaling.
4 Discussion
Our results provide evidence for an essential role of cholesterol-rich membrane domains in EDHF signaling and in agonist-induced activation of endothelial phopholipase A2. We demonstrate that reduction of membrane cholesterol results in disintegration of Cav-1- and cPLA2-containing low density membrane-domains (caveolae) that are essential for EDHF-mediated vasorelaxation. Our results suggest caveolae to play a key role in EDHF signaling.
Caveolae are considered as a specific subset of lipid rafts which are enriched in cholesterol and the cholesterol-binding scaffold protein caveolin-1 [23]. Caveolin-1 is typically expressed in vascular endothelium including the intima of pig coronary arteries [24], and caveolin-1 containing membranes have been recognized as structures that serve endocytosis and cellular trafficking [25] but also endothelial/smooth muscle communication [23]. In particular, the control of endothelial NO production by caveolin-1 which is part of an eNOS containing signaling module in caveolae is well documented [26]. Here, we present the first evidence for a key role of caveolae in the communication between the endothelium and vascular smooth muscle via the EDHF phenomenon. Cholesterol scavenging with MβCD, a well established experimental strategy to disrupt cholesterol-rich membrane structures such as caveolae [22,27], resulted in a significant reduction of the non-NO-/non-prostacyclin-mediated relaxation of pig coronary arteries initiated by bradykinin. Several components of the signaling cascade that is activated by bradykinin in endothelial cells have been proposed to reside in caveolae. G-protein coupled receptors, including bradykinin receptors [28], as well as GTP-binding proteins and phospholipase C [29] are likely targeted to caveolar signaling complexes and may therefore represent a target of cholesterol depletion. However, we observed that IP3-mediated mobilization of Ca2+ from intracellular stores was not affected by cholesterol depletion and the EDHF-responses initiated by receptor-independent mobilization of intracellular Ca2+ using ionomycin were also inhibited by MβCD. These results suggest that inhibition of bradykinin-induced EDHF signaling by cholesterol depletion was not based on disruption of receptor- or G-protein function and pointed to a target of inhibition distal to cellular IP3 production. Since caveolae have been proposed to host a number of proteins involved in cellular Ca2+ signaling [18,27,30], we studied the intracellular Ca2+ signals associated with Ca2+ entry that is triggered by depletion of intracellular Ca2+ stores. The results obtained in classical Ca2+ re-addition protocols strongly argue against inhibition of Ca2+ entry mechanisms or disturbance of other endothelial Ca2+ transport systems as the mechanism of suppression of EHDF signaling by MβCD. In search of a cholesterol-dependent signaling system that is involved in EDHF generation, we studied the cellular localization and function of Ca2+-sensitive phospholipase A2, which produces arachidonic acid, a putative precursor molecule in EDHF signaling. Arachidonate metabolites have repeatedly been proposed to serve endothelium dependent hyperpolarization of vascular smooth muscle [9,20] and a key role of cPLA2 in the EDHF phenomenon has consistently been proposed [21]. With the present study, we provide two lines of evidence for targeting of endothelial cPLA2 to caveolae. First, using a cell fractionation protocol designed to isolate caveolae, we observed a significant co-fractionation of cPLA2 and caveolin-1 in low density membrane fractions, and the localization of the two proteins on density gradients was highly sensitive to cholesterol extraction. Secondly, co-immunoprecipitation experiments suggested physical interaction between cPLA2 and Cav-1, the scaffold protein of caveolae. Thus, a fraction of cellular cPLA2 is targeted to cholesterol and caveolin-1-rich membranes. This result is in line with a recent report which demonstrates that PLA2 interacts with caveolin-1 in neuronal cells, suggesting a role of caveolin-1 in hormone-induced activation of PLA2. Association of cPLA2 with the scaffold domain of caveolin-1 has been suggested as an important factor for suppression of cellular PLA2 activity [12]. It is therefore tempting to speculate about a relationship between subcellular targeting, caveolin-binding and activity of endothelial phopholipase A2. Stimulation of endothelial cells with bradykinin, indeed resulted in reduced association of cPLA2 in caveolin-1-containing complexes. This finding is consistent with the observed inhibitory interaction between caveolin-1 and neuronal PLA2. Cholesterol depletion resulted in significant distortion of the cellular localization of caveolin-1 and cPLA2, but not in dissociation of cPLA2–Cav-1 complexes. Thus, the effects of MβCD on PLA2 regulation and EDHF signaling do not involve dissociation of caveolin-1 from PLA2 containing complexes, but may be based on altered cellular localization and changes in the lipid environment of PLA2. Taken together, our results are in line with the concept that Ca2+-dependent cPLA2 is targeted to caveolae and regulated as recently proposed for neuronal cells [12]. At present, we cannot exclude that caveolae host additional signaling components involved in the EDHF signaling distal to Ca2+-dependent liberation of arachidonic acid. Nonetheless, our data strongly suggest disturbance of cPLA2 regulation as the basis of MβCD-induced inhibition of EDHF signaling. It is of note that EDHF-signaling displays substantial quantitative heterogeneity among the vascular tree. Although EDHF was reported to play a minor role in endothelial—smooth muscle communication of macrovessels such as aorta, expression of putative EDHF synthases, which utilize arachidonate as a substrate, has been demonstrated for pig aortic endothelial cells [31]. Thus, this cell system appears suitable to study EDHF signaling. The observed MβCD-induced reduction of Ca2+-dependent arachidonic acid generation may be due to decreased steady-state substrate availability as a consequence of disrupted caveolae targeting and enhanced constitutive activity of cPLA2. We suggest that the endothelial arachidonic acid metabolism that is involved in EDHF generation occurs in caveolae and is thus sensitive to cholesterol depletion.
In aggregate, our results demonstrate that EDHF signaling in pig coronary arteries involves Ca2+-dependent cPLA2, an enzyme that is able to form a complex with caveolin-1 and resides in low-density, caveolin-1 containing membranes, i.e. caveolae. We show that disintegration of caveolae with MβCD is associated with suppression of EDHF signaling, an effect that is blunted by supplementation of arachidonate. These data strongly support the concept of a pivotal role of caveolin-rich lipid rafts in vascular physiology and pathophysiology. We propose that caveolae govern EDHF-mediated endothelial/smooth muscle communication due to a key role in cellular regulation of endothelial cPLA2 activity. The integrity of caveolae is suggested as a prerequisite for coordinated and effective control of vascular tone of the endothelium. The pathophysiological significance of alterations in caveolin-dependent EDHF signaling remains to be clarified.
Acknowledgments
Supported by the Austrian Science Funds (P-14950 and F715 to KG and P16860-B9 and F714 to WFG).
Abbreviations
- PAEC
pig aortic endothelial cells
- DEA/NO
2-(N, N-diethylamino)-diazenolate-2-oxide.diethylammonium salt
- EDHF
endothelium-derived hyperpolarizing factor
- PBS
phosphate buffered saline
- U46619
9, 11-dideoxy-11α, 9α-epoxymethano-prostaglandin F2α
- l-NNA
Nω-nitro-l-arginine
- MβCD
methyl-beta-cyclodextrin
- cPLA2
cytosolic calcium-dependent phospholipase A2
- dpm
disintegrations per minute
References
Author notes
Time for primary review 25 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}