





Abstract
CaMKII is a serine–threonine protein kinase that is abundant in myocardium. Emergent evidence suggests that CaMKII may play an important role in promoting atrial fibrillation (AF) by targeting a diverse array of proteins involved in membrane excitability, cell survival, calcium homeostasis, matrix remodelling, inflammation, and metabolism. Furthermore, CaMKII inhibition appears to protect against AF in animal models and correct proarrhythmic, defective intracellular Ca2+ homeostasis in fibrillating human atrial cells. This review considers current concepts and evidence from animal and human studies on the role of CaMKII in AF.
1. Introduction
Atrial fibrillation (AF) is the most common sustained clinical arrhythmia, affecting approximately 33.5 million people annually worldwide.1 Age is a major risk factor for AF, and the disease incidence and prevalence have increased with the ageing of the general population.2,3 AF is associated with increased morbidity, primarily from stroke and heart failure, and increased mortality.4,5
AF is a progressive disease, and the current clinical classification of AF is based on its pattern and duration.6 AF is classified as paroxysmal AF (episodes that last <7 days with or without intervention), persistent AF (episodes lasting ≥7 days), long-standing persistent AF (continuous AF >12 months in duration), or permanent AF (when no further attempts are made to achieve normal sinus rhythm). Affected individuals do not necessarily progress through these various forms of AF in a linear or predictable fashion. The pathophysiology of AF consists of mechanisms involved in the initiation of the arrhythmia, maintenance of the arrhythmia, and progression to long-lasting forms of AF.7 The progression of AF is associated with the concept of ‘AF begets AF’, in which changes in atrial structure and function promote and perpetuate atrial arrhythmias,8 and these changes are referred to as atrial arrhythmogenic remodelling.9
An understanding of the molecular basis for these AF-related changes and how they pertain to the initiation and progression of the disease is essential to the development of effective therapeutic strategies. A conceptual framework for a mechanistic understanding of these mechanisms can be divided into three components.10 The first component is the basic arrhythmia mechanism responsible for the initiation and maintenance of AF. This refers to triggered (ectopic) and reentrant activities. AF initiation fundamentally requires a trigger in the form of either triggered (ectopic) activity or reentrant activity usually in the presence of a vulnerable substrate that may be anatomic or functional. An exception will be in lone AF in which AF occurs in the absence of structural heart disease. Both focal ectopic firing and reentrant mechanisms can maintain AF; however, the latter is the likely mechanism in the majority of cases. The second mechanistic component is atrial arrhythmogenic remodelling that promotes AF and comprises electrical, structural, and autonomic nervous system and Ca2+-handling changes observed in AF.9 The last component consists of the aetiological factors that predispose to AF. These include concurrent cardiovascular diseases such as heart failure, sinus node dysfunction (SND), valvular heart disease, hypertension, and ageing; genetic factors such as single gene mutations, gene polymorphisms, and gene variants; and other extrinsic factors such as sleep apnoea, obesity, and thyroid disease. Atrial remodelling can occur either as a consequence of these aetiological factors or as a direct consequence of AF itself.
The Ca2+- and calmodulin-dependent protein kinase II (CaMKII) is a multifunctional serine–threonine kinase that is now recognized as a critical Ca2+ and reactive oxygen species (ROS) sensor that mechanistically links several of the observed cellular perturbations and mechanisms in AF with proarrhythmic atrial remodelling.
The goal of this review is to highlight new and emerging evidence highlighting the role of CaMKII in AF and how CaMKII participates in the complex interplay of the various mechanistic components of AF. We start with an overview of CaMKII and relevant downstream targets and then proceed to discuss known and potential effects of CaMKII in the spectrum of AF disease. We conclude with a discussion of potential therapeutic implications. Most of our knowledge regarding CaMKII comes from studies performed in ventricular cardiomyocytes. Although discussing a wide range of studies, we have tried to emphasize a growing body of work in humans and animals using atrial myocardium.
2. CaMKII—biology, activation, and downstream targets
2.1 Biology and structure
CaMKII is a multifunctional serine–threonine protein kinase that is abundantly expressed in various tissues, including the heart. There are four CaMKII gene products resulting in four homologous isoforms of CaMKII—α, β, δ, and γ, and these share activation mechanisms.11–14 These isoforms are thought to co-assemble as heteromultimeric holoenzymes and are known to have varied expression levels in different tissues12 (Figure 1). For instance, CaMKIIα and CaMKIIβ are preferentially enriched in neuronal tissues, whereas CaMKIIδ and CaMKIIγ are the predominant isoforms in the heart. There is a hypervariable region in CaMKII located between the association domain and the C-terminus of the regulatory domain that gives rise to various splice variants. There is a CaMKIIδ splice variant with a nuclear localization signal (NLS; CaMKIIδB) and another lacking an NLS sequence (CaMKIIδC).15 The presence or absence of the NLS does not absolutely determine localization, perhaps because these isoforms co-assemble as heteromultimers and the ratio of the NLS-containing isoforms biases cellular targeting.15 Nevertheless, the specific role of these splice variants remains unclear, and both CaMKIIδB and CaMKIIδC are present in nuclear and cytosolic compartments.16
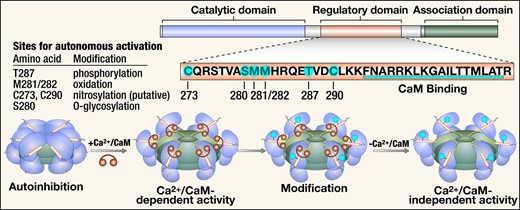
Structure, activation, and post-translational modifications of CaMKII. CaMKII monomers consist of a catalytic domain, a regulatory domain, and an association domain. The regulatory domain has an autoinhibitory binding region with several sites for post-translational modification and a CaM binding region. Co-assembly of monomers through the association domain forms the CaMKII holoenzyme. In the presence of Ca2+, Ca2+/CaM binds to the regulatory site, which causes a conformational change and activation of the autoinhibited inactive enzyme with exposure of the catalytic domain. Post-translational modification of the autoinhibitory region at any of the sites highlighted results in constitutive activity of CaMKII that is autonomous of Ca2+/CaM. CaM, calmodulin.
Structurally, CaMKII exists as a holoenzyme that consists of two stacked ring-shaped hexamers.14 Each monomer consists of an N-terminal catalytic domain, a central regulatory domain, and a C-terminal association domain.17 The catalytic domain harbours the adenosine triphosphate (ATP) and substrate binding sites and is responsible for the catalytic activity of the protein. The regulatory domain contains an inhibitory pseudosubstrate sequence, several amino acid residues susceptible to post-translational modification, and the calmodulin (CaM) binding region. The association domain is responsible for binding with other subunits to form the holoenzyme (Figure 1).14
2.2 Activation
In the inactive state of CaMKII, the pseudosubstrate region of the regulatory domain inhibits the catalytic domain of each CaMKII subunit by sterically blocking the substrate and ATP-binding pocket.18,19 With increases in intracellular [Ca2+], Ca2+ binds to CaM and the calcified CaM (Ca2+/CaM) activates CaMKII by binding to the C-terminal region of the regulatory domain. This causes a conformational change with allosteric displacement of the pseudosubstrate region, allowing the substrate and ATP access to the catalytic domain.20 Sustained increases in Ca2+/CaM in the presence of ATP result in autophosphorylation at Thr 287 in the autoinhibitory region of the regulatory domain. Autophosphorylation promotes a conformational change that prevents the association of the catalytic and regulatory domains, resulting in sustained Ca2+-independent activation.21–23
Our group discovered that oxidation of a pair of methionine residues at positions 281/282 results in a similar form of Ca2+/CaM-independent persistent activation compared with autophosphorylation.24 Other modes of post-translational modification that result in autonomous CaMKII activity include O-linked glycosylation of serine at position 280 by O-linked N-acetylgluocasmine25 and NO-dependent nitrosylation of cysteine residues at putative positions 116, 273, or 290.26 Interestingly, phosphorylation of Thr 306/307 causes CaMKII inactivation by decreasing the binding to Ca/CaM complexes.27 Very recently, it was discovered that NO modification of Ser 273, a position outside of the autoinhibitory domain, inactivates CaMKII.28 Thus, CaMKII may be activated or inactivated by post-translational modifications, but post-translational modifications to the CaMKII autoinhibitory domain provide a path for CaMKII to become persistently active under the influence of upstream signals that increase intracellular [Ca2+] and ROS.24,29
The capacity of CaMKII to transduce the effects of a multitude of upstream messengers raises the question of the existence of a central mechanism that regulates the activation of CaMKII similar to the protein kinase A (PKA) signalling pathway. Such a central mechanism does not appear to exist; rather what is clear is that initiation of CaMKII activation requires Ca2+/CaM binding, and sustained elevation in intracellular [Ca2+] can promote Ca2+/CaM autonomous activity by ‘autophosphorylation’ at Thr 287. However, Ca2+/CaM-independent activity can also follow increased ROS, even in the presence of physiological (diastolic) intracellular [Ca2+].30 Additionally, similar to PKA, β-adrenergic stimulation can activate CaMKII through the cAMP-Epac pathway (discussed subsequently). Changes in global cystolic [Ca2+], such as those occurring in excitation–contraction coupling (ECC) and local [Ca2+], for example, in the dyadic space can activate CaMKII. CaMKII achieves signal specificity through a variety of mechanisms, as discussed subsequently.
2.3 Localization and intracellular targeting
Cellular signalling systems generally have mechanisms that facilitate the interaction of effector molecules with their targets often through close proximity to achieve signal specificity and efficiency. CaMKII achieves subcellular targeting in a heterogeneous fashion such as compartmentalization, employing anchoring proteins and also identifying target protein motifs with homology to the CaMKII regulatory domain autoinhibitory region.11,31 CaMKII is enriched in specific subcellular domains such as those in the transverse tubules adjacent to l-type Ca2+ channels (LTCC) and ryanodine receptors.32 Earlier we briefly mentioned the existence of CaMKIIδ splice variants—CaMKIIδB and δC. It was initially thought that CaMKIIδB preferentially localized to the nucleus and CaMKIIδC to the cytosol; however, it has subsequently been shown that these isoforms are not exclusively limited to these cellular compartments.16 In fact, these and other isoforms of CaMKII co-assemble as heteromultimers, and the relative composition of the heteromultimer biases cellular targeting. The study of Mishra et al.16 showed that there is compartmentalized activation of CaMKIIδ independent of the isoform with the location of activation determined by the stimulus and the Ca2+ release site. Phenylephrine preferentially activated both CaMKIIδ isoforms in the nuclear compartment, whereas caffeine preferentially activated these isoforms in the sarcoplasmic reticulum (SR). Compartmentalization also had functional significance, with phenylephrine selectively phosphorylating nuclear transcriptional regulator HDAC5 at a specific CaMKII site (Ser 498) and caffeine selectively phosphorylating phospholamban (PLN) at the CaMKII-specific phosphorylation site (Thr 17).
Unlike other signalling molecules such as PKA that have extensively validated anchoring proteins that act as adaptors, there is less known about how CaMKII achieves target specificity. However, a few anchoring proteins have been described that regulate coupling of CaMKII to some target proteins, thus enhancing signalling specificity. The first of these was the N-methyl-d-aspartate subunit NR2B in the nervous system, which caused activation-dependent subcellular targeting through translocation.33 Subsequently, other anchoring proteins have been described in the heart. Alpha-kinase anchoring protein is an example of a protein that anchors sarcoplasmic endoplasmic reticulum ATPase (SERCA2a) and CaMKII, and through this interaction modulates CaMKII-dependent PLN phosphorylation.34 Another is β-arrestin that interacts with CaMKIIδ35 and functions as an anchoring protein that links β1-adrenergic receptors to the cAMP-Epac-CaMKII signalling pathway.36 It forms a multimeric complex with CaMKII and Epac in the cytoplasm, and this complex then translocates to the plasma membrane after β-adrenergic stimulation to bring CaMKII and Epac in close proximity with cAMP. A membrane-associated guanylate kinase protein, SAP97, has also been shown in rat atrial myocytes to anchor CaMKII to the C-terminus of Kv4.3 orchestrating CaMKII-mediated increases in the transient outward current (Ito).37
CaMKII uses recognition and targeting of CaMKII-binding motifs in its interaction with LTCC by binding to a CaMKII binding site on the β2a subunit of LTCC.38 Similarly, CaMKII uses this mechanism to bind to βIV-spectrin in targeting voltage-gated Na+ channels.39 Taken together, CaMKII employs different mechanisms as described to achieve target specificity and efficiency in its role as a master regulator of several cellular processes.
2.4 Downstream targets
CaMKII acts on diverse downstream targets in cardiomyocytes by lowering the free energy required to phosphorylate a target serine or threonine residue. Phosphorylation can lead to conformational changes that affect protein function. A full discussion of CaMKII target proteins is beyond the scope of this review, but CaMKII targets related to myocardial ECC may be particularly important for understanding the role of CaMKII in AF. CaMKII appears to affect myocardial cell membrane excitability through its action on most voltage-gated ion channels such as LTCCs,40–42 K+ channels,37,43,44 and Na+ channels.45–47 Other CaMKII targets include Ca2+ cycling proteins such as the type 2 ryanodine receptor (RyR2)48 and phospholamban (PLN).49 The net effect of CaMKII phosphorylation on these targets, in the context of pathological stress, is to promote a proarrhythmic circumstance by favouring cell membrane hyperexcitability and afterdepolarizations. Collectively, early afterdepolarizations (EADs) and delayed afterdepolarizations (DADs) are thought to be triggers for initiating arrhythmias, including AF (see the section on Mechanisms and experimental models of AF).
2.4.1 l-type Ca2+ channels
The LTCC is the major portal for extracellular Ca2+ to enter the cytoplasm.50 CaMKII-catalysed phosphorylation of LTCCs produces high-activity mode-2 gating, resulting in increased open probability of ICa.L channels.38 This increases the influx of Ca2+ into atrial myocytes. CaMKII also results in ‘Ca2+-dependent ICa.L-facilitation’40 in which there is increased peak ICa.L and slowed ICa.L inactivation,51 a signature LTCC response to repeated depolarizing pulses.11 Mode-2 gating, and ICa facilitation are thought to be proarrhythmic by predisposing to action potential (AP) prolongation, EADs, SR Ca2+-leak, inward Na+/Ca2+ exchanger (NCX) current, and DADs.32,38,52,53
2.4.2 Voltage-gated Na+ channels
The α isoform of the cardiac sodium (Na) channel (Nav1.5) is phosphorylated by CaMKII.45 This leads to Na+ gating modification with increased steady-state inactivation of the Na2+ channel with high heart rates. CaMKII-dependent phosphorylation of Nav1.5 also slows Na2+ current (INa) inactivation and augments the non-inactivating ‘late’ component of INa.39,54 Sustained late INa increases subsarcolemmal [Na+] and indirectly increases intracellular [Ca2+] by prolonging AP duration (APD) and by reducing the efficacy of the NCX to extrude Ca2+ from the cytoplasm to the extracellular space.55 These effects contribute to EADs and DADs. CaMKII inhibition by autocamide-2-related inhibitory peptide in murine and human atrial myocytes or genetic knockout of CaMKIIδc (CaMKIIδc-knockout mice) prevents late INa-dependent SR Ca2+-leak.56
2.4.3 Voltage-gated K+ channels
Voltage-gated K+ (IK) currents are primarily responsible for membrane repolarization. IK is a composite of the effect of several ion channels. Here, we discuss the IK channels that are best understood to be directly affected by CaMKII. The transient outward K+ current (Ito) is responsible for early repolarization and termination of the plateau phase of the AP, and CaMKII activates Ito in atrial myocytes.43,57 This results in slowing of Ito inactivation and acceleration of its recovery, with the overall effect of shortening the AP and decreasing the refractory period. The KV4.3 pore-forming subunit of Ito is regulated by CaMKII-dependent phosphorylation through the accessory protein SAP97, and this also results in increased Ito.37 KV1.4 is postulated to have a CaMKII phosphorylation target motif.58 The inward rectifier current (IK1) is another component of IK that is activated by CaMKII.46 This is the primary cardiac inward rectifier current, and the protein Kir2.1 subunit is the essential pore-forming unit for this channel. IK1 is important for maintaining the resting cell membrane potential and sculpting terminal phase 3 repolarization. IK1 is increased in chronic AF,59,60 and this helps sustain high-frequency reentrant sources called rotors that are characteristic of reentrant arrhythmias.61IK1 and the ultrarapid delayed rectifier K+ current (IKUR) both appear to be augmented by CaMKII,43,57 which offsets the APD-prolonging effects of CaMKII-dependent ICa.L and INa phosphorylation. Contrary to earlier studies, a study of atrial myocytes from human subjects in SR and AF showed that CaMKII inhibition with KN-93 had no effect on IK1 current in sinus rhythm or chronic AF patients, whereas protein kinase C (PKC) decreased IK1 current in chronic AF.62 Further, PKC-dependent phosphorylation also increased constitutive acetylcholine-sensitive K+ current (IK.Ach) activity in chronic AF.62 Thus, complex actions at multiple voltage-gated ion channels can contribute to arrhythmias by promoting EADs and DADs (i.e. arrhythmia triggers) and by augmenting dispersion of repolarization that may be a substrate for reentry.
2.4.4 Type 2 ryanodine receptor
The RyR2 is an intracellular Ca2+ channel that controls release of myofilament activating Ca2+ from the SR lumen to the cytoplasm. This Ca2+ initiates the mechanical phase of ECC, but can also influence Ca2+-sensitive cell membrane conductances (i.e. by actions on ion channels, exchangers, and transporters), transcription, and Ca2+-regulated mitochondrial processes. A mostly LTCC-mediated trigger of intracellular Ca2+ is the fundamental signal for activating RyR2, but its opening probability is biased by phosphorylation. CaMKII-dependent hyperphosphorylation of Ser 2814 on RyR2 increases open probability, augmenting dyssynchronous SR Ca2+ release48 and proarrhythmic DADs,63 which are mostly due to inward NCX current (INCX). There is increased NCX expression in persistent AF, which magnifies the size of DAD-generating inward INCX for any given amount of aberrant Ca2+ release. RyR2 dysfunction that destabilizes the closed state increases AF susceptibility in genetic mouse models.64–66 Inhibition of CaMKII-dependent RyR2 phosphorylation reduces the incidence of rapid pacing-induced AF in mice.66,67
2.4.5 Phospholamban
Phospholaman (PLN) is a negative regulator of the SERCA2a.68,69 Phosphorylation of PLN by PKA (at Ser 16) or CaMKII (Thr 17) promotes PLN aggregation and disengagement from SERCA2a and SERCA2a disinhibition, resulting in increased SR Ca2+-reuptake from the cytoplasm.70,71
Activation of these target proteins, by CaMKII-catalysed phosphorylation, can explain the acute proarrhythmic role of CaMKII in AF and other arrhythmias, by enhancing the probability of afterdepolarizations and reentry.72,73 Chronic CaMKII activation favours cardiomyocyte death and fibrosis, myocardial hypertrophy, and extracellular matrix remodelling—processes that contribute to proarrhythmic remodelling of the tissue substrate. In the case of AF, tissue substrate remodelling is an important factor in the transition of AF from sporadic or paroxysmal forms to chronic and permanent forms.72,74
Regulation of phosphorylation, and thus activity of the various Ca2+-handling proteins discussed earlier, does not depend solely on the actions of CaMKII. Other protein kinases such as PKA and PKC also play significant roles in the regulation of these proteins. For the purposes of this review, we briefly highlight the PKA signalling pathway and how this interacts with CaMKII signalling in the context of AF. Both PKA and CaMKII act at different sites on these proteins with similar consequences in most, but not all, cases. Some of the known PKA phosphorylation sites for these proteins include Ser 1700 for LTCC, Ser 2030 and Ser 2808 for RyR2, and Ser 16 for PLN. Classic PKA signalling involves cyclic adenosine monophosphate (cAMP) activation of PKA in response to β-adrenergic signalling. PKA activation under physiological conditions occurs through β-adrenergic signalling and causes intracellular influx of Ca2+ and increased SR Ca2+ uptake and storage that culminates in increased systolic Ca2+ transients.75 CaMKII activation occurs either directly through increased intracellular Ca2+ (as discussed earlier) or alternatively through a cAMP-dependent mechanism. The latter occurs via activation of adenylyl cyclases with subsequent increase in cytosolic cAMP that then binds and activates a guanine nucleotide exchange protein activated by cAMP (Epac). Epac modulates cardiac Ca2+-handling in various ways such as increasing the frequency of Ca2+ oscillations and inducing SR Ca2+ release independent of PKA through effectors such as phospholipase C and CaMKII.76–78 PKC also may affect CaMKII through this mechanism.78 Epac increases CaMKII-mediated diastolic Ca2+-leak through RyR2 activation.77 This is considered the cAMP- and Epac-induced SR Ca2+-leak pathway. RyR2 PKA hyperphosphorylation is present in atrial tissue from a canine model of atrial pacing-induced AF and in atrial tissue from humans with AF when compared with sinus rhythm,79 and this results in increased open probability of the channel.80
β-adrenergic stimulation also induces PKA-dependent and PKA-independent CaMKII-mediated diastolic SR Ca2+-leak.80,81 Late INa also increases diastolic SR Ca2+-leak in atrial myocytes/myocardium from mice and humans with permanent AF by CaMKII- and PKA-dependent phosphorylation of PLN (Thr 17 and Ser 16, respectively) and RyR2 (Ser 2814 and Ser 2808, respectively).56 Both kinases appeared to be necessary for late INa-dependent SR Ca2+-leak induction.
It is clear that several signalling pathways including cAMP/PKA, cAMP/Epac, and direct CaMKII activation are involved in AF; however, the relative contribution of PKA and CaMKII and the relevant signalling pathways to the initiation, maintenance, or progression of AF are unclear. Previous studies suggest differential contribution of both kinases. For instance, PKA activity significantly activated ICa and SR Ca2+ uptake with modest effects on RyR2.82,83 CaMKII, in contrast, had more robust RyR2 effects with modest ICa activation and SR Ca2+ uptake.84 Historically, there has been some controversy on the effects of PKA and CaMKII on RyR2, with CaMKII considered to be downstream of PKA. However, more recent studies demonstrate that these kinases act independently but synergistically on β-adrenergic stimulation.56,81,85 There is ample evidence of crosstalk between PKA and CaMKII-dependent signalling at different levels. With regard to Ca2+-signalling, CaMKII affects cAMP levels in a negative-feedback loop fashion through its action on phosphodiesterase 4D, thus indirectly affecting PKA activity.86 PKA also facilitates CaMKII-dependent SR Ca2+-leak induction by maintaining SR Ca2+-load.56
3. CaMKII in AF
CaMKII appears to be a myocardial stress response modulator because its activity and expression are increased in diverse forms of structural heart disease. Furthermore, CaMKII activity seems to augment classical fight or flight heart rate responses.87Tables 1 and 2 show a summary of various studies evaluating CaMKII protein expression and activity in animal models (Table 1) and human tissue (Table 2) with atrial tachyarrhythmias and AF.
CaMKII in AF: summary of animal studies
| Author | Year | Species | Arrhythmia model | CaMKII | Effect |
|---|---|---|---|---|---|
| Greiser et al.88 | 2009 | Goat | Rapid atrial pacing | CaMKII-dependent hyperphosphorylation of RyR2 | Atrial hypocontractility Decreased SR Ca2+ load |
| Chelu et al.89 | 2009 | Mouse | Genetic mouse model (CREM-IbΔC-X transgenic mice) | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2, arrhythmia suppression by CaMKII inhibition. |
| Chelu et al.89 | 2009 | Goat | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2 |
| Wakili et al.90 | 2010 | Canine | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Atrial hypocontractility Decreased APD Decreased Ca2+ transient |
| Purohit et al.66 | 2013 | Mouse | Angiotensin-II | Increased ox-CaMKII | Increased AF susceptibility dependent on ox-CaMKIIδ and RyR2 S2814 phosphorylation, arrhythmia suppression by CaMKII inhibition |
| Greiser et al.91 | 2014 | Rabbit | Atrial myocytes | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Fischer et al.56 | 2015 | Mouse | Atrial cardiomyocytes treated with ATX-II | Increased p-CaMKII | Enhanced late INa Increased SR Ca2+-leak Hyperphosphorylation of CaMKII target sites: RyR2 (Ser2815) and PLN (Thr17) |
| Lenski et al.92 | 2015 | NVRM | Electrical stimulation | Increased p-CaMKII/CaMKII | Increased p-AMPK/AMPK Increased FA uptake Decreased glucose uptake |
| Author | Year | Species | Arrhythmia model | CaMKII | Effect |
|---|---|---|---|---|---|
| Greiser et al.88 | 2009 | Goat | Rapid atrial pacing | CaMKII-dependent hyperphosphorylation of RyR2 | Atrial hypocontractility Decreased SR Ca2+ load |
| Chelu et al.89 | 2009 | Mouse | Genetic mouse model (CREM-IbΔC-X transgenic mice) | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2, arrhythmia suppression by CaMKII inhibition. |
| Chelu et al.89 | 2009 | Goat | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2 |
| Wakili et al.90 | 2010 | Canine | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Atrial hypocontractility Decreased APD Decreased Ca2+ transient |
| Purohit et al.66 | 2013 | Mouse | Angiotensin-II | Increased ox-CaMKII | Increased AF susceptibility dependent on ox-CaMKIIδ and RyR2 S2814 phosphorylation, arrhythmia suppression by CaMKII inhibition |
| Greiser et al.91 | 2014 | Rabbit | Atrial myocytes | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Fischer et al.56 | 2015 | Mouse | Atrial cardiomyocytes treated with ATX-II | Increased p-CaMKII | Enhanced late INa Increased SR Ca2+-leak Hyperphosphorylation of CaMKII target sites: RyR2 (Ser2815) and PLN (Thr17) |
| Lenski et al.92 | 2015 | NVRM | Electrical stimulation | Increased p-CaMKII/CaMKII | Increased p-AMPK/AMPK Increased FA uptake Decreased glucose uptake |
AP, action potential; AMPK, AMP-activated protein kinase; ATX-II, Anemonia-sulcata toxin-II; FA, fatty acid; NVRM, neonatal ventricular rat myocytes; ox-CaMKII, oxidized CaMKII; p-CaMKII, phosphorylated CaMKII; p-AMPK, phosphorylated AMPK; PLN, phospholamban; SR, sarcoplasmic reticulum; RyR2, ryanodine type 2 receptor.
CaMKII in AF: summary of animal studies
| Author | Year | Species | Arrhythmia model | CaMKII | Effect |
|---|---|---|---|---|---|
| Greiser et al.88 | 2009 | Goat | Rapid atrial pacing | CaMKII-dependent hyperphosphorylation of RyR2 | Atrial hypocontractility Decreased SR Ca2+ load |
| Chelu et al.89 | 2009 | Mouse | Genetic mouse model (CREM-IbΔC-X transgenic mice) | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2, arrhythmia suppression by CaMKII inhibition. |
| Chelu et al.89 | 2009 | Goat | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2 |
| Wakili et al.90 | 2010 | Canine | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Atrial hypocontractility Decreased APD Decreased Ca2+ transient |
| Purohit et al.66 | 2013 | Mouse | Angiotensin-II | Increased ox-CaMKII | Increased AF susceptibility dependent on ox-CaMKIIδ and RyR2 S2814 phosphorylation, arrhythmia suppression by CaMKII inhibition |
| Greiser et al.91 | 2014 | Rabbit | Atrial myocytes | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Fischer et al.56 | 2015 | Mouse | Atrial cardiomyocytes treated with ATX-II | Increased p-CaMKII | Enhanced late INa Increased SR Ca2+-leak Hyperphosphorylation of CaMKII target sites: RyR2 (Ser2815) and PLN (Thr17) |
| Lenski et al.92 | 2015 | NVRM | Electrical stimulation | Increased p-CaMKII/CaMKII | Increased p-AMPK/AMPK Increased FA uptake Decreased glucose uptake |
| Author | Year | Species | Arrhythmia model | CaMKII | Effect |
|---|---|---|---|---|---|
| Greiser et al.88 | 2009 | Goat | Rapid atrial pacing | CaMKII-dependent hyperphosphorylation of RyR2 | Atrial hypocontractility Decreased SR Ca2+ load |
| Chelu et al.89 | 2009 | Mouse | Genetic mouse model (CREM-IbΔC-X transgenic mice) | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2, arrhythmia suppression by CaMKII inhibition. |
| Chelu et al.89 | 2009 | Goat | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Hyperphosphorylation of RyR2 |
| Wakili et al.90 | 2010 | Canine | Rapid atrial pacing | Increased expression and autophosphorylation of CaMKIIδ | Atrial hypocontractility Decreased APD Decreased Ca2+ transient |
| Purohit et al.66 | 2013 | Mouse | Angiotensin-II | Increased ox-CaMKII | Increased AF susceptibility dependent on ox-CaMKIIδ and RyR2 S2814 phosphorylation, arrhythmia suppression by CaMKII inhibition |
| Greiser et al.91 | 2014 | Rabbit | Atrial myocytes | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Fischer et al.56 | 2015 | Mouse | Atrial cardiomyocytes treated with ATX-II | Increased p-CaMKII | Enhanced late INa Increased SR Ca2+-leak Hyperphosphorylation of CaMKII target sites: RyR2 (Ser2815) and PLN (Thr17) |
| Lenski et al.92 | 2015 | NVRM | Electrical stimulation | Increased p-CaMKII/CaMKII | Increased p-AMPK/AMPK Increased FA uptake Decreased glucose uptake |
AP, action potential; AMPK, AMP-activated protein kinase; ATX-II, Anemonia-sulcata toxin-II; FA, fatty acid; NVRM, neonatal ventricular rat myocytes; ox-CaMKII, oxidized CaMKII; p-CaMKII, phosphorylated CaMKII; p-AMPK, phosphorylated AMPK; PLN, phospholamban; SR, sarcoplasmic reticulum; RyR2, ryanodine type 2 receptor.
CaMKII in AF: summary of human studies
| Author | Year | Species | Type of AF | Tissue | CaMKII | Effect |
|---|---|---|---|---|---|---|
| Tessier et al.43 | 1999 | Human | Chronic AF | RA | Increased CaMKIIδ expression | CaMKII slowed Ito inactivation; CaMKII inhibition hastened Ito inactivation |
| Christ et al.93 | 2004 | Human | Chronic AF | RAA | Increased protein expression of the catalytic subunit of protein phosphatase 2A and phosphatase activity | Decreased basal ICaL density in chronic AF |
| Chelu et al.89 | 2009 | Human | Chronic AF | RAA | Increased CaMKIIδ expression and autophosphorylation | Hyperphosphorylation of RyR2 |
| Neef et al.94 | 2010 | Human | Unclassified | RA | Increased CaMKII expression | Increased SR Ca2+-leak and diastolic Ca Increased RyR2 phosphorylation, reversal of SR Ca2+-leak with CaMKII inhibition |
| Voigt et al.95 | 2012 | Human | Chronic AF | RAA | Increased autophosphorylation of CaMKII | Hyperphosphorylation of RyR2 Increased SR Ca2+-leak, diastolic Ca2+, frequency of spontaneous Ca2+ release events Reversal of SR Ca2+-leak and afterdepolarizations with CaMKII inhibition |
| Purohit et al.66 | 2013 | Human | Mixed (paroxysmal, persistent/permanent, and unclassified) | RA | Increased ox-CaMKII | AF patients had more ox-CaMKII compared with patients in sinus rhythm |
| Greiser et al.91 | 2014 | Human | Persistent AF | RA | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Christ et al.96 | 2014 | Human | Chronic AF | RAA | Reduced CaMKII-dependent phosphorylation of PLN but not RyR2 | Blunting of agonist-evoked arrhythmias in atrial myocytes from patients with AF compared with those in sinus rhythm |
| Lenski et al.92 | 2015 | Human | Permanent AF | LAA | Increased p-CaMKII/CaMKII | Increased FA uptake Decreased glucose uptake |
| Author | Year | Species | Type of AF | Tissue | CaMKII | Effect |
|---|---|---|---|---|---|---|
| Tessier et al.43 | 1999 | Human | Chronic AF | RA | Increased CaMKIIδ expression | CaMKII slowed Ito inactivation; CaMKII inhibition hastened Ito inactivation |
| Christ et al.93 | 2004 | Human | Chronic AF | RAA | Increased protein expression of the catalytic subunit of protein phosphatase 2A and phosphatase activity | Decreased basal ICaL density in chronic AF |
| Chelu et al.89 | 2009 | Human | Chronic AF | RAA | Increased CaMKIIδ expression and autophosphorylation | Hyperphosphorylation of RyR2 |
| Neef et al.94 | 2010 | Human | Unclassified | RA | Increased CaMKII expression | Increased SR Ca2+-leak and diastolic Ca Increased RyR2 phosphorylation, reversal of SR Ca2+-leak with CaMKII inhibition |
| Voigt et al.95 | 2012 | Human | Chronic AF | RAA | Increased autophosphorylation of CaMKII | Hyperphosphorylation of RyR2 Increased SR Ca2+-leak, diastolic Ca2+, frequency of spontaneous Ca2+ release events Reversal of SR Ca2+-leak and afterdepolarizations with CaMKII inhibition |
| Purohit et al.66 | 2013 | Human | Mixed (paroxysmal, persistent/permanent, and unclassified) | RA | Increased ox-CaMKII | AF patients had more ox-CaMKII compared with patients in sinus rhythm |
| Greiser et al.91 | 2014 | Human | Persistent AF | RA | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Christ et al.96 | 2014 | Human | Chronic AF | RAA | Reduced CaMKII-dependent phosphorylation of PLN but not RyR2 | Blunting of agonist-evoked arrhythmias in atrial myocytes from patients with AF compared with those in sinus rhythm |
| Lenski et al.92 | 2015 | Human | Permanent AF | LAA | Increased p-CaMKII/CaMKII | Increased FA uptake Decreased glucose uptake |
Ito, transient outward K+ current; RA, right atrium; RAA, right atrial appendage; LAA, left atrial appendage; FA, fatty acid; ox-CaMKII, oxidized CaMKII; p-CaMKII, phosphorylated CaMKII; SR, sarcoplasmic reticulum; PLN, phospholamban; RyR2, ryanodine type 2 receptor.
CaMKII in AF: summary of human studies
| Author | Year | Species | Type of AF | Tissue | CaMKII | Effect |
|---|---|---|---|---|---|---|
| Tessier et al.43 | 1999 | Human | Chronic AF | RA | Increased CaMKIIδ expression | CaMKII slowed Ito inactivation; CaMKII inhibition hastened Ito inactivation |
| Christ et al.93 | 2004 | Human | Chronic AF | RAA | Increased protein expression of the catalytic subunit of protein phosphatase 2A and phosphatase activity | Decreased basal ICaL density in chronic AF |
| Chelu et al.89 | 2009 | Human | Chronic AF | RAA | Increased CaMKIIδ expression and autophosphorylation | Hyperphosphorylation of RyR2 |
| Neef et al.94 | 2010 | Human | Unclassified | RA | Increased CaMKII expression | Increased SR Ca2+-leak and diastolic Ca Increased RyR2 phosphorylation, reversal of SR Ca2+-leak with CaMKII inhibition |
| Voigt et al.95 | 2012 | Human | Chronic AF | RAA | Increased autophosphorylation of CaMKII | Hyperphosphorylation of RyR2 Increased SR Ca2+-leak, diastolic Ca2+, frequency of spontaneous Ca2+ release events Reversal of SR Ca2+-leak and afterdepolarizations with CaMKII inhibition |
| Purohit et al.66 | 2013 | Human | Mixed (paroxysmal, persistent/permanent, and unclassified) | RA | Increased ox-CaMKII | AF patients had more ox-CaMKII compared with patients in sinus rhythm |
| Greiser et al.91 | 2014 | Human | Persistent AF | RA | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Christ et al.96 | 2014 | Human | Chronic AF | RAA | Reduced CaMKII-dependent phosphorylation of PLN but not RyR2 | Blunting of agonist-evoked arrhythmias in atrial myocytes from patients with AF compared with those in sinus rhythm |
| Lenski et al.92 | 2015 | Human | Permanent AF | LAA | Increased p-CaMKII/CaMKII | Increased FA uptake Decreased glucose uptake |
| Author | Year | Species | Type of AF | Tissue | CaMKII | Effect |
|---|---|---|---|---|---|---|
| Tessier et al.43 | 1999 | Human | Chronic AF | RA | Increased CaMKIIδ expression | CaMKII slowed Ito inactivation; CaMKII inhibition hastened Ito inactivation |
| Christ et al.93 | 2004 | Human | Chronic AF | RAA | Increased protein expression of the catalytic subunit of protein phosphatase 2A and phosphatase activity | Decreased basal ICaL density in chronic AF |
| Chelu et al.89 | 2009 | Human | Chronic AF | RAA | Increased CaMKIIδ expression and autophosphorylation | Hyperphosphorylation of RyR2 |
| Neef et al.94 | 2010 | Human | Unclassified | RA | Increased CaMKII expression | Increased SR Ca2+-leak and diastolic Ca Increased RyR2 phosphorylation, reversal of SR Ca2+-leak with CaMKII inhibition |
| Voigt et al.95 | 2012 | Human | Chronic AF | RAA | Increased autophosphorylation of CaMKII | Hyperphosphorylation of RyR2 Increased SR Ca2+-leak, diastolic Ca2+, frequency of spontaneous Ca2+ release events Reversal of SR Ca2+-leak and afterdepolarizations with CaMKII inhibition |
| Purohit et al.66 | 2013 | Human | Mixed (paroxysmal, persistent/permanent, and unclassified) | RA | Increased ox-CaMKII | AF patients had more ox-CaMKII compared with patients in sinus rhythm |
| Greiser et al.91 | 2014 | Human | Persistent AF | RA | No change in CaMKII-dependent RyR2 phosphorylation or CaMKII activity | Ca2+ signalling silencing with increased intracellular Ca2+ and no changes in Ca2+ sparks, Ca2+ waves, Ca2+ release, and Ca2+ release instability |
| Christ et al.96 | 2014 | Human | Chronic AF | RAA | Reduced CaMKII-dependent phosphorylation of PLN but not RyR2 | Blunting of agonist-evoked arrhythmias in atrial myocytes from patients with AF compared with those in sinus rhythm |
| Lenski et al.92 | 2015 | Human | Permanent AF | LAA | Increased p-CaMKII/CaMKII | Increased FA uptake Decreased glucose uptake |
Ito, transient outward K+ current; RA, right atrium; RAA, right atrial appendage; LAA, left atrial appendage; FA, fatty acid; ox-CaMKII, oxidized CaMKII; p-CaMKII, phosphorylated CaMKII; SR, sarcoplasmic reticulum; PLN, phospholamban; RyR2, ryanodine type 2 receptor.
4. Mechanisms and experimental models of AF
AF and atrial tachycardia-related remodelling are the consequences of a complex interplay of pathophysiological mechanisms. An understanding of the fundamental processes that constitute the molecular basis of AF is necessary to highlight the particular steps now thought to be affected by CaMKII. The primary mechanisms of AF are focal ectopic or triggered activity and reentrant activity.97 Triggered activity can occur after full repolarization of the AP and these are called DADs. Alternatively, EADs are triggered activities that occur during phase 2 or 3 of the AP. Ca2+ overload with associated shortening of the APD favours generation of DADs and these are rate-dependent, whereas EADs are favoured by APD prolongation, persistent inward ICa.L,32INa,39 and spontaneous SR Ca2+ release.98–100 EADs and DADs can also be driven by adrenergic stimulation, at least in part because of their action to activate CaMKII and its downstream targets. Reentrant activity occurs in the presence of suitable substrate, which can be either anatomic (e.g. fibrosis) or functional (e.g. electrical refractoriness) that allows for the perpetuation of an excitable circuit. The substrate can be the consequence of AF-related atrial remodelling or the result of other conditions such as genetic mutations, ageing, structural heart disease, or other systemic disease.7 Reentrant activity is the primary mode for AF maintenance. Persistent AF is likely maintained by the combination of complex multiple circuit reentry and ectopic activity.101
Importantly, as discussed in the previous section, excessive CaMKII activation has been implicated in AF triggering and reentry mechanisms through its downstream effects on ion channels and proarrhythmic remodelling that results from cell death, fibrosis, and hypertrophy.
Various experimental paradigms have been developed to study the mechanisms of AF.97 Clinical AF is a heterogeneous mix of phenotypes. AF often exists in association with structural heart disease such as congestive heart failure, valvular disease, or hypertension; however, there are times that AF occurs in the absence of any detectable structural heart disease (‘lone AF’). Also there are different clinical forms of AF: paroxysmal, persistent, and permanent AF. The dominant paradigm for the natural progression of the disease is from paroxysmal to permanent AF. The rapid atrial pacing (RAP) model8,102 of experimental AF aims to mimic ‘lone AF’. Of note, earlier studies used a single atrial burst pacemaker,103 and these were associated with high ventricular rates that likely caused mild tachycardia-mediated cardiomyopathy that potentially complicated the observations in these studies. More recent studies combine RAP with complete atrioventricular block104,105 to avoid or minimize this effect. Both models are relevant to human disease as the former simulates AF with no rate control and the latter AF with adequate rate control.97 The RAP model is also of clinical significance as a model for studying the transition from paroxysmal to chronic AF. Another AF experimental model to study the relationship between heart failure and AF exists, in which rapid ventricular pacing induces heart failure.104 There are differences in the electrical and structural remodelling observed in RAP model when compared with the heart failure model. Other forms of experimental AF in animals include AF combined with structural heart disease,106 atrial dilatation without heart failure,107 and sterile pericarditis to mimic post-operative AF.108 Increasingly, transgenic mice are being used as AF experimental models.66,89,109,110 The main advantage of mouse models is their genetic tractability and ability to generate mutant genes to mimic human diseases and to dissect candidate AF molecular mechanisms. For the purposes of this review, we will focus primarily on the RAP model in ‘large’ animals, as a model to study AF-related atrial remodelling and AF progression, and on various genetically modified mouse models with the power to dissect defined molecular components of AF pathways.
The transition from paroxysmal to the more chronic forms of AF is poorly understood, and the role that CaMKII plays in this process is an area of on-going interest in the field. The most established role for CaMKII in AF in in vivo (animal) systems relates to mechanisms of arrhythmia triggering, by contributing to RyR2 hyperphosphorylation and Ca2+-leak, which triggers afterdepolarizations.56,67,89,111 Importantly, this mechanism pertains to fibrillating human atrium.94,95 Although triggering may be important for paroxysmal or more permanent forms of AF,95 it appears to be required for paroxysmal AF in the absence of structural heart disease (i.e. lone AF). This is consistent with reports in which CaMKII contributes to triggering in pulmonary vein cardiomyocytes.112 Taken together, these data align with the idea that CaMKII contributes to early stage AF. However, CaMKII could also contribute to the transition from paroxysmal to more permanent forms of AF by several mechanisms: loss of normal intracellular [Ca2+] homeostasis, increased cardiomyocyte death and fibrosis, matrix remodelling by elaboration of matrix metalloproteinase-9 (MMP-9), and inflammatory gene transcription. Although intriguing and of potential clinical relevance, the role of CaMKII in late stage AF pathophysiology is less completely understood.
5. Components of atrial remodelling in AF
Atrial remodelling is characterized by several atrial structural and functional alterations that occur either as a result of AF or underlying structural heart disease that predates or coexists with AF. These changes can be classified as electrical remodelling, tissue (structural) remodelling, Ca2+-handling abnormalities, and neurohormonal.9 Although tissue, Ca2+, and electrical remodelling are complex and involve diverse signalling pathways, CaMKII has emerged as a nodal signal with the potential to orchestrate many of these changes. Furthermore, CaMKII activity is activated downstream to neurohumoral agonists, suggesting that CaMKII is a transduction signal linking neurohumoral signalling and ROS with proarrhythmic mechanisms in AF. Here, we will focus on the components that are known or postulated to be directly modulated by CaMKII and also discuss several CaMKII-dependent molecular changes that occur in AF.
6. Electrical remodelling
Electrical remodelling entails changes in the electrophysiological properties of the atria. This is a composite of the alterations that occur in ion channels, pumps, and exchangers in atrial cells.
APD shortening and enhanced variability of APD are central features of AF-related electrical remodelling and are accompanied by loss of rate adaptation of the atrial effective refractory period (AERP), which facilitates the maintenance of reentrant circuits in both animal AF models and human AF.8,113–115 Reduced AP duration is a result of reduced ICa,L.113,116,117 The reduced ICa,L measured in atrial myocytes isolated from chronic AF patients may indicate an adaptive response to arrhythmia-induced intracellular Ca2+ overload.117 The reduction in ICa,L is due to decreased mRNA and protein expression of the α subunit of ICa,L(CaV1.2) in AF through the negative feedback action of Ca2+-dependent phosphatase calcineurin/nuclear factor of activated T-cells (NFAT).118 Translocation of NFAT into the nucleus results in transcriptional downregulation of the CaV1.2 gene with consequent AP duration shortening.118 Although most of the available data show increased mRNA and protein levels of CaV1.2, others have found no alteration in protein levels in AF.93,119 An intriguing finding in one of these studies was that CaMKII inhibition with KN-93 resulted in about a 40% reduction in basal ICa,L in patients with sinus rhythm, but had no effect on basal ICa,L in patients with chronic AF.93 This was associated with increased phosphatase activity (especially protein phosphatase 2A) in chronic AF, suggesting that an imbalance in the kinase-phosphatase activity in favour of increased phosphatase activity is responsible for the reduced basal ICa,L in patients with chronic AF. The local CaMKII activity was not measured, but hypothetically there could be a local reduction in CaMKII activity due to several factors, such as cytoskeletal protein abnormalities due to AF that affect CaMKII activity, medications, or differences in experimental models.93,119 Also, increased activation of the Ca2+-dependent protease calpain promotes breakdown of ICa,L channels.120 Taken together, this suggests that the net effect on ICa,L may depend more on post-translational modification of the channel by phosphorylation rather than by expression.
A recent study by Greiser et al.91 challenges the current paradigm in the field that atrial remodelling results from dysfunctional Ca2+ handling that results in increased triggered activity and the role that CaMKII plays in tachycardia-mediated atrial remodelling. They observed a paradoxical suppression of central Ca2+ release with no changes in Ca2+ sparks, Ca2+ waves, SR Ca2+ release, and Ca2+ release instability in atrial myocytes from rabbits exposed to RAP for 5 days and in atrial myocytes from chronic AF patients. This was termed ‘Ca2+ signalling silencing’. It was attributed to increased Ca2+ buffering capacity of the atrial myocytes through binding to myofilaments and reduction in RyR2 expression with smaller RyR2 clusters in the SR. Additionally, there was hyperphosphorylation of RyR2 at the PKA phosphorylation site (Ser 2808), but reduced CaMKII-dependent RyR2 phosphorylation (Ser 2815) with no changes in CaMKII and PLN activity. It should be noted that the animal model used was a short-lived duration (5 days) of atrial pacing, with no evidence of AF in the rabbits. Further studies are needed to determine whether Ca2+ signalling silencing is a transient response to tachycardia or a more sustained response to chronic rapid activation with potential relevance to the pathophysiology of AF. Another group has reported a marked reduction in agonist-induced arrhythmias in the atria of patients with AF when compared with those in sinus rhythm with a corresponding reduction in maximal ICaL responses.96 The agonists used were norepinephrine, epinephrine, serotonin, and forskolin (a cAMP activator). CaMKII inhibition with KN-93 reduced norepinephrine-evoked ICaL responses in the atria from myocytes from patients with sinus rhythm but not in atrial myocytes from patients with AF. Also, adrenergic stimulation evoked reduced CaMKII-dependent phosphorylation of PLN (Thr 17), but no changes in PKA-dependent PLN phosphorylation (Ser 16) and no changes in RyR2 phosphorylation either by CaMKII or by PKA. Taken together, this was suggestive of loss of CaMKII-dependent phosphorylation of LTCC and PLN in response to adrenergic stimulation in chronic AF. It is not clear to us how to reconcile these results from Christ with the report by Voigt et al.,95 showing that CaMKII is critical for agonist-evoked RyR2 Ca2+-leak and arrhythmia triggering. These studies highlight the complexity of the mechanisms involved in AF and more specifically raise a question about the role of CaMKII in chronic AF. Further investigations to definitively understand these issues better are needed.
Decreased atrial conduction velocity may contribute to reentry and maintenance of AF. This occurs via a reduction in INa or direct inhibition of gap-junction channels in atrial cardiomyocytes.121 In murine experiments, decreased INa was shown to be due to an acute Ca2+-dependent inhibition of INa and reduced Nav1.5 subunit expression under chronic conditions.122 This Ca2+-dependent reduction in INa promotes the stability of AF primarily by a reentrant mechanism, but may also reduce ectopic activity.123 Oxidized CaMKII (ox-CaMKII) slows conduction in the infarct border zone of ventricular myocardium,124 but it is currently unknown whether CaMKII modulates atrial conduction velocity. However, it has been suggested that CaMKII-dependent phosphorylation could also reduce INa, especially at fast heart rates as seen in AF.45
7. Structural remodelling
The hallmark of structural atrial remodelling is atrial fibrosis.104,125 It is a common final pathway for AF promoting conditions and a negative predictor of treatment outcomes.9,126 Atrial fibrosis results from interaction between atrial cardiomyocytes and fibroblasts. Ca2+ influx into atrial fibroblasts induces proliferation and differentiation into collagen-secreting myofibroblasts, resulting in increased fibrosis and consequently heterogeneous conduction slowing and reentry.127 These changes are brought about by influx of Ca2+ into atrial fibroblasts via multiple ion channels. In human atrial fibroblasts, transient-receptor potential (TRP) channels are major participants in this process, especially TRP melastatin-related-7 (TRPM7) and canonical-3 (TRPC3) channels.128,129 CaMKII putatively can act both upstream and downstream of these TRP channels.130 AF patients have larger TRPM7 currents in their atrial fibroblasts, and knockdown of TRPM7 expression reduces basal differentiation of fibroblasts from chronic AF patients.128 It is unknown whether CaMKII interacts with TRPM7 in atrial myocytes, although CaMKII was shown to inhibit TRPM7-like channels in hepactocytes.131 Influx of Ca2+ through TRPC3 channel promotes CaMKII activation and NADPH-oxidase-mediated production of ROS in a genetic mouse model of dilated cardiomyopathy.132
CaMKII is involved in profibrotic signalling in the myocardium. Aldosterone stimulation leads to increased ox-CaMKII and activation of myocyte enhancer factor 2 that causes increased myocardial MMP-9 synthesis.29 Angiotensin II increases ox-CaMKII that causes sinoatrial cell death, SND, and atrial fibrosis, whereas CaMKII inhibition is protective against death and fibrosis in sinoatrial cells.133 Further investigation is needed to determine whether CaMKII contributes to atrial fibrosis in AF by these or other pathways, but CaMKII inhibition does reduce fibrosis in ventricular myocardium.29
Excessive CaMKII activity triggers cell death, and this results in reparative fibrosis and adverse remodelling in ventricular myocytes in a pressure overload model of heart failure,134 ventricular myocytes, and sinoatrial cells following Ang II infusion.24,133 CaMKII inhibition is also protective against beta-adrenergic-receptor agonist-induced apoptosis, and this anti-apoptotic mechanism involves PLN regulation of SR Ca2+ content135,136 and mitochondria.137 Mitochondrial-targeted CaMKII inhibition prevents myocyte death following myocardial infarction and catecholamine infusion137 (discussed subsequently). These various CaMKII-mediated mechanisms of cell death have not been directly tested in AF.
There is an established link between inflammation and AF,138,139 and several inflammatory markers such as cytokines, C-reactive protein, and complementary factors are elevated in AF. Inflammation leads to structural remodelling, primarily in the form of fibrosis. We previously showed that CaMKII induces local inflammatory response through the nuclear factor-κB pathway in cardiomyocytes when exposed to bacterial endotoxin or myocardial infarction and CaMKII inhibition suppressed this response.140 CaMKII is also oxidatively activated during myocardial infarction or by endotoxin as part of a toll-like receptor/myeloid differentiation protein 88 pathway.141 All these findings suggest that CaMKII may play an important role in proarrhythmic atrial remodelling favouring AF.
Atrial enlargement is another component of structural remodelling. Mice with transgenic overexpression of the transcriptional repressor CREM-IbΔC-X in cardiomyocytes (CREM mice) develop age-dependent progression from spontaneous atrial ectopy to paroxysmal and long-lasting AF episodes.142 Ca2+-handling abnormalities and atrial enlargement precede the spontaneous development of AF. Genetic inhibition of CaMKII-dependent RyR2 phosphorylation, by knockin replacement of a validated CaMKII-targeted phosphorylation site (RyR2-Ser 2814), in CREM mice prevents Ca2+-handling abnormalities and spontaneous AF, atrial dilatation, and conduction abnormalities.142 Thus CaMKII-dependent RyR2 hyperphosphorylation leads to both triggered activity and progressive development of an AF substrate, promoting atrial hypertrophy and dilatation, and AF progression. These mechanistic insights suggest that targeted treatment of CaMKII or SR Ca2+-leak via RyR2 has the potential to slow or arrest the progression of AF.
8. Role of CaMKII in ROS/redox sensing and signalling
ROS are involved in modulating the action of various cellular targets with functional consequences under physiological and pathological conditions in the cardiovascular system. AF is associated with increased oxidative stress, and this has been linked to AF-related atrial remodelling.143–145 There is increasing interests in the role of ROS and CaMKII in cardiac disease, with significant progress made in recent years in our understanding of links between ROS and CaMKII in the pathophysiological mechanisms of AF.146
ROS directly activates CaMKII by oxidation with subsequent persistent constitutive CaMKII activity, which is proarrhythmic in its effect on downstream targets.24,66,147 The acute proarrhythmic actions of ox-CaMKII are due to increased EADs and DADs54,148 and reentry.124
NADPH oxidases (Nox) are important sources of ROS in AF.149 Increased Nox4 expression in cardiomyocytes is associated with mitochondrial damage and apoptosis.150 In a zebrafish model, overexpression of Nox is associated with an arrhythmogenic phenotype, and this effect is mediated by ROS-induced activation of CaMKII.151 Mice that lack functional Nox (p47−/− mice) are resistant to angiotensin II-dependent increases in ROS and oxidation of CaMKII and are also resistant to angiotensin-II-mediated AF.66 Inhibition of Nox4 by apocynin attenuates electrical remodelling and AF inducibility in a rabbit model of RAP.152 As the duration of AF increases, sources of intracellular ROS shift from Nox to other cellular sources, including mitochondrial oxidases (xanthine oxidase), monoamine oxidase, and uncoupled eNOS.153
In a computational model,154 the arrhythmogenic pattern of EADs observed during oxidative stress was dependent on ox-CaMKII activation and pacing-cycle length. Oxidative stress is also associated with reduced AERP.143
Despite the link between oxidative stress and AF, attempts at utilizing antioxidants to prevent AF and AF-related atrial remodelling have been largely disappointing.155 One idea is that general antioxidants lack potency, pharmacokinetic attributes, or access to key subcellular domains necessary to exert beneficial actions. The shift from Nox as a source of cellular ROS to other cellular sources may explain why statins, which decrease ROS production by Nox via inhibition of Rac1, are only effective in acute AF models such as post-operative AF but ineffective in ameliorating ROS production in chronic AF models such as permanent AF.153 There was a recent comprehensive review of this topic.153 The finding that genetically modified mice with ROS-resistant CaMKII were protected from AF triggered by rapid pacing and primed by angiotensin II infusion66 suggests that CaMKII could be part of a concise ROS-responsive molecular pathway and a candidate therapeutic antioxidant target for AF.
9. CaMKII and mitochondria, energy expenditure, and its implications for AF mechanism
Mitochondria are essential for ATP production, the primary form of myocardial cellular energy, and they are the primary sources of ROS. They are an integral component of intracellular Ca2+ and ROS signalling. In cardiomyocytes, excessive mitochondrial Ca2+ and ROS cause opening of the mitochondrial permeability transition pore (mPTP), which leads to dissipation of the mitochondrial inner membrane potential and decreased ATP synthesis and this results in myocardial cell death via apoptotic mechanisms.156–158 CaMKII through its dual action of increasing ROS production and intracellular Ca2+ via SR Ca2+-leak results in mPTP opening.159 We previously showed that CaMKII is present in the mitochondria and that cardiomyocytes from transgenic mice with mitochondrial-targeted CaMKII inhibitory protein137 had a blunted mPTP response to Ca2+. These mice were protected from programmed cell death with exposure to different myocardial stressors such as ischaemia/reperfusion, myocardial infarction, and adrenergic stimulation.137 At this time, the constituents of mPTP are unknown and this pathway has not been directly tested in AF models. CaMKII also increases mitochondrial Ca2+ by phosphorylating the mitochondrial Ca2+ uniporter,137 although this is controversial.160 This Ca2+-sensitive channel is a major Ca2+ entry portal through the mitochondrial inner membrane into the mitochondria.161–163 Thus, mitochondrial CaMKII may contribute to a feed-forward mechanism by which Ca2+ overload triggers mPTP opening and increases ROS production, thereby causing more CaMKII activation and activity.
Increased oxidative stress and increased ox-CaMKII have been demonstrated in the atria of AF patients,66,145,164 and this was associated with mitochondrial DNA damage and reduction in ATP synthesis in these patients.164 Additionally, mitochondria undergo structural and morphological changes such as swelling and disturbance of cristae structure during RAP and AF.164–166 Functionally, impaired oxidative phosphorylation and respiration are observed in atrial tissue in response to RAP.166,167 These changes were Ca2+-dependent and were prevented by blocking LTCCs.
Ca2+ thus functions as a second messenger that links AF to structural and functional mitochondrial changes. Also, CaMKII plays an important role in the pathological mitochondrial changes that result in mitochondrial dysfunction and myocardial cell death in AF and likely contributes to atrial remodelling. The potential role of mitochondrial CaMKII represents an interesting and potentially important frontier in CaMKII research in AF.
10. CaMKII and atrial metabolic remodelling in AF
There is interplay between CaMKII and AMP-activated protein kinase (AMPK). AMPK is a serine/threonine kinase that regulates cellular energy metabolism and protein synthesis.168 CaMKII, Ca2+/calmodulin-dependent protein kinase kinase (CaMKKII), or Ca2+ alone can modulate the activity of AMPK in its role as a cellular energy sensor.169,170 The net effect of AMPK activation is enhancing energy-generating pathways and reducing energy demand processes.168,171,172 Lenski et al.92 recently demonstrated both in vitro and in vivo that arrhythmia induces metabolic changes in atrial myocardium characterized by lipid accumulation and reduced glucose uptake. They postulated that activation of AMPK causes these changes and CaMKII can activate AMPK. Inhibition of CaMKII and AMPK independently prevented these changes in vitro. This pathway involves elevated fatty acid translocase (FAT/CD36) membrane expression with resultant increased fatty acid uptake and lipid accumulation. Also, there is decreased membrane expression of synaptosomal-associated protein 23 in the plasma membrane, which results in reduced AMPK-induced glucose transporter subtype 4 membrane translocation and glucose uptake.92 These changes correlated with activation of proapoptotic pathways and atrial remodelling.
AMPK activation in AF was recently shown in canine and human atrial myocytes to be protective against AF-induced metabolic stress.173 High atrial rate and associated metabolic stress resulted in decreased ICa.L, intracellular Ca2+ transients, and cell shortening. The increased energy demand caused AMPK phosphorylation and subsequent regulation of Ca2+ handling through activation of and interaction with intracellular targets such as Cav1.2 and NCX, to counteract the effects of the metabolic stress. There was no evidence of interaction with other Ca2+-handling proteins such as RyR2, PLN, and SERCA2a. AMPK phosphorylation was increased in canine left atrial tissue after 1 week of tachypacing model of AF. Similarly, fractional AMPK phosphorylation was increased in human right atrial appendage tissue from subjects with paroxysmal AF compared with SR; however, there was a significant decrease in chronic AF subjects. This finding is interesting as it may be an indication that the AMPK system protects against paroxysmal or short bouts of AF, but does not necessarily play a role in persistent AF. It also buttresses the idea that there are likely different mechanisms underlying the different clinical forms of AF.
11. Atrial mechanical dysfunction
Atrial mechanical dysfunction, which manifests as atrial hypocontractility, is another feature of AF-related atrial remodelling and is a major cause of stroke and thromboembolism associated with AF.174,175 It coexists with on-going AF and can persist for variable periods after conversion to sinus rhythm in patients with long-standing AF.176 The latter is known as ‘atrial stunning’, and the degree of hypocontractility correlates with the duration of AF.176 In a canine atrial tachycardia model, there was reduced fractional cell shortening in atrial myocytes, and this was explained, in part, by abnormal intracellular Ca2+ handling and decreased systolic Ca2+ transients.175,177 In humans with prolonged AF, atrial contractility is reduced by as much as 75%.177 Others have shown that AP duration shortening could be responsible for the observed atrial hypocontractility.178 Additionally, in a canine model of tachypacing, there is altered myofilament function due to abnormal phosphorylation of myosin and myosin-binding protein C (MyBP-C).90 In this study, there was increased autophosphorylation and expression of CaMKIIδ and protein phosphatase I activity with downregulation of PKA. However, a direct link between CaMKII and altered myofilament function was not shown. Several studies show that MyBP-C is phosphorylated by CaMKII, which leads to hypocontractility and impaired relaxation in heart failure.179–181 Acute β-adrenergic stimulation leads to CaMKII phosphorylation of MyBP-C,179 but the effect of chronic β-adrenergic stimulation on this and the functional significance is unclear.182 Titin, which is a large myofilament protein, contributes to myocardial diastolic stiffness, and its mechanical properties are changed in heart failure.183–185 Specific CaMKII phosphorylation sites were recently identified on titin.186 CaMKII-dependent titin phosphorylation resulted in decreased myocardial passive force and altered diastolic stress in murine and human failing hearts,186 potentially linking diastolic dysfunction and AF via CaMKII. Thus, CaMKII acts through diverse mechanisms with the potential to reduce mechanical function. However, more definitive studies and more thorough identification of key CaMKII target sites will be necessary to test these concepts definitively.
12. AF and heart failure
AF commonly coexists with structural heart disease such as heart failure in patients.187,188 Atrial CaMKII activity is increased in a ventricular tachypacing-induced canine model of heart failure189 and in goats with atrial dilatation.88 In heart failure, there is chronic activation of neurohormonal agonist signalling, which begins as an adaptive response but subsequently becomes pathological and contributes to disease progression. This chronic pathological response results from increased intracellular Ca2+ and ROS.11 The agonists involved in this pathway are β-adrenergic receptors agonists, angiotensin II, and aldosterone, and all activate CaMKII.24,29,190 Indeed, CaMKII transcription, protein expression, and activity are increased in the failing heart.29,191–193 In addition to the neurohormonal system activation, other mechanisms by which CaMKII is increased in heart failure include increased calcineurin activity.194,195 The mechanism of increased CaMKII activity in heart failure appears to hinge on autophosphorylation of Thr 287 and/or oxidation of methionines 281 and 282.11 We found that ox-CaMKII was increased after myocardial infarction and was further enhanced by angiotensin II,24 aldosterone,29 or hyperglycaemia.196 Pathological β-adrenergic receptor stimulation in heart failure could activate CaMKII by cAMP-EPAC-dependent and cAMP-independent mechanisms to hyperphosphorylate RyR, causing SR Ca2+-leak and arrhythmias.77,78,85 As described earlier, the downstream effects of CaMKII activation likely result in AF-related atrial remodelling changes that promote AF initiation and progression. This may explain, in part, the association between heart failure and AF.
13. SND and AF
AF frequently coexists with SND.197 In the MOST trial, among patients with SND who required pacemakers, AF was the most frequent arrhythmia. About 45–50% of the subjects had a history of AF at the time of pacemaker implantation.198 SND is associated with changes in the sinoatrial pacemaker complex that are similar to AF-related atrial remodelling.199,200 AF has been shown to cause SND201 and conversely, SND predisposes to AF.199,202,203 The mechanisms for these observations are due to atrial remodelling—structural remodelling especially fibrosis,204 electrical remodelling, and neurohormonal stimulation, as described earlier in this review.
CaMKII through its effect on intracellular Ca2+ dynamics plays a key role in pacemaker activity.205 In this study, inhibition of CaMKII either by autocamide-2-inhibitory peptide (AIP), a specific peptide inhibitor, or by KN-93 resulted in decreased ICa.L amplitude and complete arrest of sinoatrial cells. Work from our laboratory indicated that CaMKII contributes approximately half of the dynamic range of fight or flight heart rate increases by actions in sinoatrial cells32 and that angiotensin II infusion in mice caused increased atrial and sinoatrial node ox-CaMKII. Ox-CaMKII triggered a critical threshold of sinoatrial cell apoptosis, atrial fibrosis, and conduction slowing that contributed to SND.133 Targeted CaMKII inhibition in the heart or sinoatrial node of mice protected against the deleterious effects of angiotensin II: sinoatrial cell apoptosis, fibrosis, and SND. We also found elevated levels of ox-CaMKII in the right atria from human patients with SND133 and AF when compared with those in sinus rhythm and also in the atria of Ang-II-infused mice.66 These mice had increased ROS, SR Ca2+ sparks, DADs, and heightened AF susceptibility compared with saline-infused mice. Mice with genetically engineered resistance to CaMKII oxidation and mice lacking functional NADPH oxidase (p47−/−) were resistant to angiotensin-II-mediated AF. These data show that ox-CaMKII is involved in the pathophysiology of both AF and SND and may be important in the clinical coexistence and synergy between both diseases.
14. Genetics and CaMKII in AF
It is recognized that there is a genetic contribution to AF, and certain familial and inheritable forms of AF have been described. Additionally, common genetic variants and single nucleotide polymorphisms have been identified in relation to AF. This was recently comprehensively reviewed.206 Although CaMKII has been implicated in some inherited arrhythmias such as catecholaminergic polymorphic ventricular tachycardia (CPVT), long QT type 3, ankyrin-B syndrome (long QT type 4),207,208 and Timothy syndrome (long QT type 8),209 we are unaware of a specific CaMKII genetic mutation or variation that has been identified in relation to AF. A loss-of-function mutation in ANK2 (a gene that encodes for ankyrin-B) was noted in some patients with early onset AF.210 In this study, mice with ankyrin-B deficiency manifested electrophysiological properties consistent with human AF, and DeGrande et al.207 showed that CaMKII inhibition rescued the proarrhythmic features of ankyrin-B deficiency in models of human ankyrin-B syndrome. Also, gain-of-function mutations in RyR2 have been shown to predispose to CPVT and AF.65,89,211,212
15. Potential as a therapeutic target in AF
Current treatment strategies for the management of AF include anti-arrhythmic drugs, ablation, and surgical procedures. Although useful, these oftentimes have suboptimal efficacy, and the anti-arrhythmic drugs have potential adverse effects and toxicities, including life-threatening proarrhythmia.213 The central role of Ca2+-dysregulation and CaMKII in the pathophysiology of AF makes CaMKII an attractive therapeutic target.
15.1 Pharmacological approaches to modulation of CaMKII activity
Several CaMKII-specific inhibitors exist, but none possesses drug-like properties necessary for therapeutic purposes. This topic was recently reviewed in detail.214 Here we discuss the use of relevant CaMKII inhibitors and highlight their use in relation to AF. KN-93 is a relatively specific CaMKII inhibitor215 that is widely employed for experimental purposes214 and is the best CaMKII inhibitor available. It competitively and selectively binds to the CaM-binding site of CaMKII, allosterically blocking activation. It does not compete with ATP and so does not have an effect on the activity of autophosphorylated CaMKII. Thus, once CaMKII is activated and autophosphorylated, KN-93 is ineffective in CaMKII inhibition, irrespective of the mode of activation. It selectively inhibits CaMKII relative to other kinases such as PKA and PKC, but has been shown to also effectively inhibit CaMKI and CaMKIV,216,217 and a few other kinase targets.218 A drawback to the use of KN-93 is its off-target effects. It affects LTCC52,219 and several voltage-gated potassium channels.220 Unfortunately, these off-target actions occur at concentrations necessary for CaMKII inhibition (e.g. IC50 in myocardial lysates of ∼2.5 mM)52 and are not shared by the control drug, KN-92. Thus, it is our view that it is best to use a variety of complementary approaches to CaMKII inhibition, such as gene deletion and inhibitory peptides, in order to be confident that experimental outcomes are not due to off-target actions. This is particularly true for studies of arrhythmias, in which ion channel antagonist actions of KN-93 have the potential to achieve anti-arrhythmic actions. However, these compounds have been useful and remain a key tool in our understanding of CaMKII signalling. KN-93 has been shown in both in vitro and in vivo mouse models to prevent pacing-induced AF.89 It also decreases SR Ca2+-leak in atrial myocytes from AF patients.94,95 KN-62 is another CaMKII inhibitor that is structurally similar to KN-93 and has the same mechanism of action, but has largely been replaced by KN-93.
CaMKII inhibitory peptides, which mimic the autoinhibitory region of the CaMKII regulatory domain, include AIP and autocamide-3 inhibitor (AC3-I). These are highly selective inhibitors of CaMKII relative to PKA, PKC, and CaMKIV221,222 and have been used experimentally to study CaMKII function in cardiovascular physiology and disease. In vivo murine studies have used internalization sequences or transgenic expression. Mice with transgenic myocardial expression of AC3-I were protected from pacing-induced AF, following angiotensin II infusion.66 There is the potential for off-target effects and altered selectivity with modification to these peptides either from fusion with green fluorescent protein, myristoylation or from changes to internalization sequences to enhance specific attributes of the peptide, such as increased expression, metabolic stability, or cell permeation.87,214,223 AC3-I has the potential to inhibit protein kinase D,223 but this potential has not been validated in other studies.224
CaMKIIN is a small protein endogenous to neurons but not to myocardium that inhibits CaMKII with high specificity and potency. We have used CaMKIIN as an experimental tool that can be delivered to specific target sites through local application of adenoviral constructs, targeted transgenic expression to specific tissues, and/or intracellular compartments.87,133,137 Sinoatrial node-targeted CaMKII inhibition by local application of adenoviral construct of CaMKIIN was protective against Ang-II-induced SND in mice.133 There are no studies of these peptides in AF models to our knowledge.
The CaMKII inhibitors described earlier have all been in the context of experimental models, but highlight the potential for CaMKII-targeted therapeutics. Although there are currently no CaMKII inhibitors available clinically, several potential small molecule inhibitors for cardiovascular application are in early stages of development.
15.2 Potential therapeutic application of CaMKII inhibition
CaMKII inhibition or elimination of CaMKII-dependent RyR2 phosphorylation has been shown to protect from AF in mouse models and is also beneficial in atrial cardiomyocytes of chronic AF patients.66,67,95 Furthermore, CaMKII is a truly nodal signal, so that CaMKII inhibition has the potential to counter AF by actions relevant to its progression from paroxysmal to permanent. However, given the ubiquitous nature of CaMKII and its multifunctional role in several physiological processes, it will be important to prove that systemic CaMKII inhibition does not contribute to undesirable side effects such as reduced fertility and impaired memory.225,226 Despite these potential issues, CaMKII is dispensable, as demonstrated by the viability of CaMKII knockout mice, and is likely present in heart to maximize heart rate and mechanical performance as part of the fight or flight response. Arguably, these attributes are not of central importance to most AF patients. A clinically useful CaMKII inhibitor likely should not cross the blood–brain barrier and should possess a sufficiently wide therapeutic window to avoid potential toxicities. Allosteric or isoform-specific CaMKII inhibitors could also enhance selectivity and reduce the potential for off-target effects.227
MicroRNAs have emerged as another potential therapeutic tool in the management of human disease.228 Some microRNAs have been identified that are involved in the regulation of CaMKIIδ expression such as microRNA-145229 and microRNA-30b-5p,230 whereas several others have been identified in the transcriptional changes that occur in AF.231 Targeting these microRNAs in the heart may emerge as a unique tool managing arrhythmias and other cardiac diseases. The pathophysiological mechanisms and importance of CaMKII may vary based on experimental AF models and in different forms of clinical AF, and the effect of CaMKII inhibition in these various conditions is unclear. Pacing-induced AF in the setting of angiotensin II infusion66 may mirror post-operative AF because of the shared circumstance of neurohumoral activation and elevated ROS. Based on the link between CaMKII activation and tissue remodelling, it is possible that a CaMKII inhibitor drug could reduce progression of AF from paroxysmal to chronic or permanent forms.
Given the landscape of potential therapeutic targets, our view is that CaMKII inhibition is a promising approach.
16. Conclusion
AF and atrial remodelling in the context of AF occur through a complex interplay of several molecular and cellular mechanisms, and these are implicated in the mechanisms for the initiation, maintenance, and progression of AF. Available evidence shows that there is excessive CaMKII activity in the setting of AF. CaMKII by its actions as a Ca2+ and ROS sensor modulates several of the pathways involved in AF and atrial remodelling to promote a proarrhythmic milieu in the atrium (Figures 2 and 3). Animal studies show that inhibition of CaMKII is protective against AF. This suggests that CaMKII inhibition is a potential therapeutic target for the management of AF. Further studies are however needed to understand the mechanistic role of CaMKII in atrial remodelling and AF.
![CaMKII-mediated intracellular mechanisms of AF-related atrial remodelling. Stress signals result in increased intracellular Ca2+ and ROS that lead to increased CaMKII expression and activity. CaMKII has diverse downstream targets through which it promotes arrhythmogenesis. Its cumulative effect on ionic channels (ICa.L, INa, IK1, Ito, and INCX) is inhomogeneous AP propagation and repolarization dispersion that leads to triggered activity. It also results in increased SR Ca2+, further increasing intracellular [Ca2+]. This acts via the calcineurin/NFAT pathway to activate profibrotic pathways and induce atrial hypertrophy. Increased intracellular [Ca2+] also leads to increased mitochondrial [Ca2+], which through its action on mPTP opening causes increased ROS and CaMKII activation in the mitochondria that can cause cell death. CaMKII also acts through AMPK activation to increase lipid accumulation and to decrease glucose uptake. β-adrenergic stimulation acts through angiotensin receptors to activate NADPH oxidase (NOX) to increase intracellular ROS and to activate CaMKI via oxidation.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/109/4/10.1093_cvr_cvw002/2/m_cvw00202.jpeg?Expires=1716405852&Signature=O2bKEqeE-EUFC51XYMpBBRv1oc-XFsFxEPb~GoKm3IzD2qn928tSwirGuHhwo8eZcuMJQ8tgde7v9vAsMy5g5e4MEpxPbBNiEPxyIKf3nK0k5uJKxZgucCRTSyBMvwoQQkkICGyeyVSff6zuAIvW8IwWOYARhNFwPlHobtNshSuiZ6EQqjzuU7nqGCi8YqdrNFEQNwSb~NXKpzW7HIiEDJfv40RSfbjyyFVqBd5YCpTI3ePRiVKuQOjWnjxL71K9Fc-hvUfhPyl0h2rtx5XhKvP~Xfubw5WiZ7y7i~PxQFtscpd~D3kYNk6TuzRnD5p4wYDcAMOQ5WludhztEbWziA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CaMKII-mediated intracellular mechanisms of AF-related atrial remodelling. Stress signals result in increased intracellular Ca2+ and ROS that lead to increased CaMKII expression and activity. CaMKII has diverse downstream targets through which it promotes arrhythmogenesis. Its cumulative effect on ionic channels (ICa.L, INa, IK1, Ito, and INCX) is inhomogeneous AP propagation and repolarization dispersion that leads to triggered activity. It also results in increased SR Ca2+, further increasing intracellular [Ca2+]. This acts via the calcineurin/NFAT pathway to activate profibrotic pathways and induce atrial hypertrophy. Increased intracellular [Ca2+] also leads to increased mitochondrial [Ca2+], which through its action on mPTP opening causes increased ROS and CaMKII activation in the mitochondria that can cause cell death. CaMKII also acts through AMPK activation to increase lipid accumulation and to decrease glucose uptake. β-adrenergic stimulation acts through angiotensin receptors to activate NADPH oxidase (NOX) to increase intracellular ROS and to activate CaMKI via oxidation.
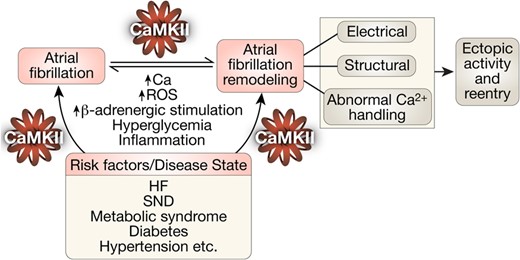
CaMKII transduces proarrhythmic signalling in atrial remodelling and AF in a feed-forward manner. Increased intracellular Ca2+ and ROS induce autonomous CaMKII activity. Excessive CaMKII activity through its action on multiple downstream targets (ion channels, proteins, etc.) results in triggered and reentrant activities that can initiate and perpetuate AF. It also results in several changes that are associated with atrial remodelling in AF. Underlying disease states or risk factors also cause excessive CaMKII activity with similar proarrhythmic downstream consequences. HF, heart failure; SND, sinus node dysfunction.
Funding
This work was supported in part by the US National Institutes of Health (NIH) grants R01-HL079031, R01-HL096652, R01-HL070250, and R01-HL113001. O.O.M. was supported by an institutional training grant from the US NIH—T32-HL007227.
Acknowledgement
We are grateful to Mr Shawn Roach who produced the artwork for the figures.
Conflict of interest: M.E.A. is a named inventor on intellectual property claiming to treat arrhythmias by CaMKII inhibition and is a cofounder of Allosteros Therapeutics, a company aiming to develop CaMKII inhibitor therapeutics. O.O.M. reports no conflict of interest.
References
Author notes
This article is part of the Spotlight Issue on Atrial Fibrillation.

{kind=link}
{kind=link}
{kind=link}