





Abstract
IFN-beta (IFNβ) signalling is increased in patients with insufficient coronary collateral growth (i.e. arteriogenesis) and IFNβ hampers arteriogenesis in mice. A downside of most pro-arteriogenic agents investigated in the past has been their pro-atherosclerotic properties, rendering them unsuitable for therapeutic application. Interestingly, type I IFNs have also been identified as pro-atherosclerotic cytokines and IFNβ treatment increases plaque formation and accumulation of macrophages. We therefore hypothesized that mAb therapy to inhibit IFNβ signalling would stimulate arteriogenesis and simultaneously attenuate—rather than aggravate—atherosclerosis.
In a murine hindlimb ischaemia model, atherosclerotic low-density lipoprotein receptor knockout (LDLR−/−) mice were treated during a 4-week period with blocking MAbs specific for mouse IFN-α/β receptor subunit 1 (IFNAR1) or murine IgG isotype as a control. The arteriogenic response was quantified using laser Doppler perfusion imaging (LDPI) as well as immunohistochemistry. Effects on atherosclerosis were determined by quantification of plaque area and analysis of plaque composition. Downstream targets of IFNβ were assessed by real-time PCR (RT-PCR) in the aortic arch. Hindlimb perfusion restoration after femoral artery ligation was improved in mice treated with anti-IFNAR1 compared with controls as assessed by LDPI. This was accompanied by a decrease in CXCL10 expression in the IFNAR1 MAb-treated group. Anti-IFNAR1 treatment reduced plaque apoptosis without affecting total plaque area or other general plaque composition parameters. Results were confirmed in a short-term model and in apolipoprotein E knockout (APOE)−/− mice.
Monoclonal anti-IFNAR1 therapy during a 4-week treatment period stimulates collateral artery growth in mice and did not enhance atherosclerotic burden. This is the first reported successful strategy using MAbs to stimulate arteriogenesis.
1. Introduction
Arteriogenesis is a natural mechanism to restore obstructed blood flow by remodelling of small, pre-existing collateral arterioles.1 In both, the coronary as well as the peripheral circulation numerous interarterial connections are present between the different vascular territories.2 Through arteriogenesis, a circulatory system arises that bypasses arterial occlusions. In days to weeks after an acute occlusion, full arterial function can be restored by arteriogenesis.3 In the presence of a sufficient collateral network, symptoms of ischaemia and the extent of myocardial infarction are reduced.4,5 Patients with coronary artery disease (CAD) show a large heterogeneity in their arteriogenic response upon coronary obstruction. In a previous study, we found up-regulation of IFN-beta (IFNβ) signalling in patients with poor coronary collateral development,6 leading to the hypothesis that IFNβ has anti-arteriogenic effects. Indeed, mice treated with IFNβ demonstrate inhibition of collateral artery growth, and mice with a genetic deletion of the IFN-α/β receptor subunit 1 (IFNAR1) show accelerated arteriogenesis.7 Thus, blockade of IFNβ signalling is a potential therapeutic target for stimulation of arteriogenesis. Interestingly, interfering with IFNβ might also have a beneficial effect on atherosclerosis. A downside of most pro-arteriogenic agents investigated in the past has been their pro-atherosclerotic properties, rendering them unsuitable for therapeutic application.8 In mouse models of atherosclerosis, IFNβ treatment accelerated lesion formation and macrophage accumulation in plaques, whereas in mice lacking myeloid IFNAR1, atherosclerosis was attenuated with a decreased accumulation of macrophages and a reduction in plaque cell death.9 We hypothesized that blocking IFNβ signalling with antibody therapy has a two-way beneficial effect in the treatment of CAD, stimulating arteriogenesis while at the same time inhibiting atherosclerosis development. Therefore, in a low-density lipoprotein receptor knockout (LDLR−/−) murine model, we determined the long-term effects of IFNAR1 MAbs on arteriogenesis as measured by laser Doppler perfusion imaging (LDPI) as well as the effects on plaque composition as determined by immunohistochemistry. An apolipoprotein E knockout (APOE−/−) model with very advanced atherosclerotic disease was used as a confirmatory model. Also, we determined the effects of IFNAR1 MAbs on macrophage polarization and expression of IFNβ downstream targets in hindlimb tissue.
2. Methods
2.1 Selection of antibody
We analysed the neutralizing capacities of three antibodies in an IFN activity assay. RAW-264.7 cells (murine monocyte–macrophage cell line; ATCC, Bethesda, MD, USA) were cultured at a density of 2 × 106 cells/mL in DMEM (Invitrogen, Bleiswijk, The Netherlands), supplemented with 10% heat-inactivated fetal bovine serum (iFBS; Lonza, Breda, The Netherlands) and 1000 U/mL of penicillin/streptomycin (Lonza). Graded concentrations of mouse IFNβ (PBL Interferonsource, Piscataway, NJ, USA) were used to stimulate the cells as indicated. We tested the capacity of the antibodies to inhibit IFNβ activity at a concentration of 2 µg/mL for each antibody: rat anti-mouse IFNβ, clone 7D3 (Abcam, Cambridge, UK); rat anti-mouse IFNβ clone RMMB-1 (PBL Interferonsource); and mouse anti-mouse IFNAR1/CD118, clone MAR1-5A3 (Leinco Technologies, St Louis, MO, USA). After 24 h of culture, total RNA was isolated from the RAW cells using the RNeasy Mini Kit (Qiagen), according to the manufacturer's protocol. IFN-response gene expression was quantified by real-time PCR. In brief, RNA was reverse-transcribed into cDNA, using the RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas, St Leon-Rot, Germany). Quantification was performed using the TaqMan Universal PCR Master Mix (Invitrogen Life Technologies, Bleiswijk, The Netherlands) with SYBR green (Invitrogen Life Technologies) as a detection agent on an ABI Prism 7900HT Fast Real-Time PCR System. mRNA expression levels of the IFN-response gene MX1 were calculated using an arbitrary standard curve and normalized to the amount of HPRT and GAPDH gene transcripts.
2.2 Primary animal experiments in LDLR−/− mice
Experiments were approved by the Institutional Animal Experimental Committee that conforms to the Guide for the Care and use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th edition, revised 2011). Female C57Bl/6 LDLR−/− were house-bred and backcrossed 10 times. At 10 weeks old, they were fed with a high-fat diet (0.15% cholesterol and 16% fat; Arie Blok, Inc., The Netherlands) for 2 months. A total of 32 LDLR−/− mice were divided into a group of 16 mice receiving antibody treatment and 16 control mice. Mice were anaesthetized by intraperitoneal (i.p.) injection of dexmedetomidine (1.0 mg/kg, Orion) and ketamine (70.0 mg/kg, Alfasan Pharma). Unilateral double ligation of the right femoral artery was performed as previously described10 and a sham operation, in which the femoral artery was dissected, was performed on the left side. Adequacy of the anaesthesia and surgical tolerance was monitored by close observation and reflex response tests. After surgery, anaesthesia was antagonized with an i.p. injection of atipamezole (0.5 mg/kg, Orion) and to minimize pain caused by surgical procedures, buprenorphine (2.0 mg/kg, MSD Animal Health) was administered subcutaneously. After ligation, the treatment group received an i.p. injection with 2.0 mg of MAbs specific for mouse IFNAR111 (MAR-1 5A3 MAb, Leinco Technologies Inc.), and control mice were injected intraperitoneally with 2.0 mg murine IgG isotype control (Leinco Technologies, Inc.). During a 4-week period, in which the high-fat diet was continued, treatment and placebo were continued twice weekly with 0.2 mg IFNAR1 MAbs and IgG isotype, respectively.
2.2.1 Laser Doppler perfusion imaging
Hindlimb perfusion was measured using LDPI (MoorLDI2, Moor Instruments, Devon, UK). LDPI was performed under anaesthesia directly before and after femoral artery ligation, as well as at 2, 7, 14, and 28 days following ligation. Mice were anaesthetized by an i.p. injection of dexmedetomidine (1.0 mg/kg, Orion) and ketamine (70.0 mg/kg, Alfasan Pharma). Excess hair from the hindlimb was removed using depilatory cream and mice were placed on a heating plate to minimize temperature variation. After LDPI, anaesthesia was antagonized with atipamezole (0.5 mg/kg, Orion). Dedicated software was used to quantify tissue perfusion. To account for variables such as ambient light, temperature, and experimental procedures, perfusion was calculated in the foot and expressed as a ratio of right (ischaemic) to left (non-ischaemic) hindlimb, as previously described.12–15
2.2.2 Histological analysis of arteriogenesis and angiogenesis
At day 28 after operation, adductor muscles were harvested. For the detection of smooth muscle cells, frozen tissue sections (5 µm) were stained for smooth muscle actin (Rabbit Anti-Mouse alpha Smooth Muscle Actin antibody, α-SMA; Abcam, 1 : 100). The antibody for CD31 (Pecam-1, M-20; Santa Cruz Biotechnology, UK, 1 : 100) was used for the detection of capillaries. Nuclei were stained with 1 : 50 000 Hoechst (Molecular Probes, Bleiswijk, The Netherlands). Images of stained sections were made with the Leica DM6000 fluorescence microscope using a ×20 objective. All analyses were done by two blinded observers using an LAS AF Lite software (Leica microsystems, IL, USA), and quantification was performed using ImageJ 1.46 (National Institutes of Health, USA).
2.2.3 Mouse blood parameters
Blood was withdrawn via tail vein incision at three time points: before start of the diet, before start of the treatment, and right before sacrifice. Plasma cholesterol and triglyceride levels were measured according to the manufacturer's protocol (Roche, Woerden, The Netherlands), and relative blood leucocyte counts were determined using the Scil Vet abc Plus analyser (Scil animal care company BV, Oostelbeers, The Netherlands).
2.2.4 Atherosclerotic lesion analysis
Mice were sacrificed for tissue isolation and further analysis after LDPI scanning at 28 days following ligation, while under anaesthesia, by cervical dislocation. Following sacrifice of the animals, the heart was dissected, frozen in Tissue-Tek (Sakura Finetek Europe BV, Alphen aan de Rijn, The Netherlands), and subsequently cut into sections of 7 µm. Serial cross-sections of every 42 µm were stained with toluidin blue, and images were obtained using the Leica DM3000 microscope (×5). Necrosis was also measured on the toluidin blue-stained slides and was identified by the presence of pyknosis, karyorrhexis, or by the complete absence of nuclei. To measure collagen, slides were air-dried, fixed in 10% formalin, washed, and then incubated for 5 min in 0.2% PMA. Hereafter, slides were incubated for 30 min in 0.05% Sirius Red (direct red 80; Sigma, Zwijndrecht, The Netherlands), which was diluted in saturated picric acid. The slides were then rinsed with 0.01 N HCl, followed by an incubation period of 5 min in this solution. Next, the slides were rinsed in a series of 70, 90, 96, and 100% ethanol followed by 2 × 3 min of xylene. Quantification of plaque size from the sections stained by Sirius Red staining was performed using the Adobe Photoshop software.
For immunohistochemistry, lesions from the aortic root were air-dried, fixed in acetone, and blocked with the Avidin/Biotin Blocking Kit (all immunohistological kits are from Vector Laboratories, Burlingame, USA; SP-2001). During the second phase of blocking, the slides were incubated with the primary antibody; 1 : 4000 MOMA-2 (macrophages, AbD Serotec, Uden, The Netherlands), 1 : 400 NIMP (neutrophils, gift from P. Heeringa), 1 : 250 KT3 (CD3, T cells, AbD Serotec), and 1 : 1000 rabbit-a-cleaved caspase-3 (apoptosis, Cell Signaling, Leiden, The Netherlands). Slides were then washed 3× with PBS and incubated for 60 min with a 1 : 300 biotin-labelled rabbit anti-rat antibody (Dako, Hervelee, Belgium) or polyvision anti-rabbit-HRP (Immunologic, Duiven, The Netherlands) for cleaved caspase-3. Hereafter, the signal was amplified using the ABC kit (except for the cleaved caspase-3 staining). Slides were washed three times with PBS and then stained using different AEC kits for MOMA-2, KT3, and cleaved caspase-3, respectively, and using Novared for NIMP. Quantification of KT3, NIMP, and cleaved caspase-3 was performed manually, whereas MOMA-2 staining was quantified using the Adobe Photoshop software.
2.2.5 Real-time PCR
Total RNA from snap-frozen aortic arches was isolated using the Qiagen RNeasy Mini Kit (Qiagen, Venlo, The Netherlands). cDNA synthesis was then performed using 500 ng of RNA with the iScript cDNA synthesis kit (BioRad, Veenendaal, The Netherlands). Real-time PCR was performed for several pro-inflammatory cytokines and chemokines, and IFNβ downstream target genes using 2 ng of cDNA, 300 nM of each primer, and 300 nmol/L of Fast SYBR Green master mix (Invitrogen Life Technologies) in a total volume of 20 µL. Forty amplification cycles were performed after which fluorescence was analysed using a computer-based imaging system (ViiA 7 Real-Time PCR system, Invitrogen Life Technologies). All gene expression levels were corrected for ARBP and cyclophilin A as housekeeping genes.
2.3 Effect of treatment at early time point
To investigate the effects of our treatment on arteriogenesis and angiogenesis, macrophage polarization, other target cells, and systemic inflammation at an early time point, we repeated our experiments in a 2-day model. Twelve female LDLR−/− were treated (six received antibody treatment and six served as controls) for 2 days following femoral artery ligation. Anaesthesia, surgical procedures including monitoring, and postoperative analgesia were performed according to the protocol of the other experiments. Mice were sacrificed after 2 days for tissue harvesting and further analysis.
2.3.1 Histological analysis of arteriogenesis, macrophages, and angiogenesis
Frozen tissue sections (5 µm) of the adductor muscle were stained to identify vessels and differentiate between M1 and M2 macrophages. Slides were fixed in dehydrated acetone for 10 min. Next, slides were blocked in PBS/1%BSA and 10% normal mouse serum. Slides were then incubated with the 1 : 80 primary rat anti-mouse F4/80 antibody (AbdSerotec, Düsseldorf, Germany) for 60 min followed by incubation with secondary antibody, Alexa Fluor 647 Goat Anti-Rat IgG (Molecular Probes). All slides were incubated overnight at 4°C with 1 : 100 primary antibody Rabbit Anti-Mouse alpha Smooth Muscle Actin antibody (α-SMA; Abcam) and with either 1 : 400 biotinylated Rat Anti-Mouse CD40 (α-CD40; eBioscience, Vienna, Austria) or 1 : 50 biotinylated Rat Anti-Mouse Mannose Receptor (α-MR; BioLegend, Cambridge, UK) for staining of M1 or M2 macrophages, respectively. Slides were incubated with secondary antibody Alexa Fluor 488 Goat Anti-Rabbit IgG (Molecular Probes) for α-SMA and Streptavidin Alexa Fluor 555 (Molecular Probes) for α-CD40 and α-MR. Nuclei were stained with 1 : 50 000 Hoechst (Molecular Probes). Angiogenesis was studied by CD31 staining and quantification of capillary density as described above.
2.3.2 Blood parameters
Blood plasma was withdrawn following sacrifice to determine the effects of anti-IFNAR1 treatment on systemic inflammation. Using flow cytometry, relative leucocyte numbers and M1 monocyte/macrophage markers on blood monocytes were determined. Blood monocytes were defined as CD45+CD11b+Ly6G− cells. Flow cytometry measurements were performed using an FACS Canto II with FACSDiva software (BD Biosciences, USA).
2.4 Animal experiments in APOE−/− mice
To confirm our results in a different model of atherosclerosis, animal experiments were repeated in 28 female C57Bl/6 APOE−/− (Charles River Laboratories, France, B6.129P2-APOE/J); of which, 14 received antibody treatment and 14 served as controls. Animals had severely advanced atherosclerosis, after being on a lipid-rich diet for 6 months, being 9 months of age at the time of sacrifice. Anaesthesia, surgical procedures including monitoring, and postoperative analgesia were performed according to the protocol of the LDLR−/− experiments. APOE−/− mice in the treatment group received an i.p. injection with 2.5 mg of IFNAR1 antibody, and control mice were injected intraperitoneally with 2.5 mg murine IgG isotype control. Antibody and IgG isotype were identical to the LDLR−/− experiments. Using methods described above, LDPI was performed before, directly after—as well as 2 and 7 days following ligation. Mice were sacrificed after 7 days for tissue harvesting and further analysis as in LDLR−/− experiments. Atherosclerotic lesion analysis was performed as in LDLR−/− animals.
2.4.1 Histological analysis of arteriogenesis, macrophages, and angiogenesis
Analysis of arteriogenesis and angiogenesis was performed on sections from calf muscle tissue. Macrophage analysis was performed on adductor muscle tissue. Protocols were used as described above.
2.4.2 Downstream targets of IFNβ in hindlimb tissue
Frozen tissue sections (8 µm) of the right adductor muscle and left adductor muscle were stained for CXCL10 and IL-27 expression. Slides were fixed in dehydrated acetone for 10 min and dried to air for 10 min. Slides were then rehydrated in EBSS/0.1% saponin (EBSS/sap) for 5 min followed by blocking with the Avidin/Biotin Blocking Kit (Vector Laboratories; SP-2001) for 15 min each and EBSS/0.1% saponin/1% block (EBSS/inc) with 10% normal mouse serum and 10% Normal Donkey Serum for 60 min. Slides were incubated overnight at 4°C with 10 µg/mL of primary antibody Goat Anti-Mouse CXCL10 (Leinco; I-236) or 10 µg/mL of primary antibody Goat Anti-Mouse IL27 (R&D Systems, Minneapolis, MN, USA; AF1834). After incubation, slides were washed 3× 5 min with EBSS/sap and incubated for 60 min with Biotinylated Donkey Anti-Goat IgG (Jackson ImmunoResearch, Baltimore, PA, USA, 705-066-147), followed by Tyramide Signal Amplification (TSA™ Kit #23, Molecular Probes/Invitrogen, T20933) including incubation with HRP-streptavidin for 90 min followed by 3× 5 min washing with EBSS/sap and incubation with Alexa Fluor 546 tyramide for 5 min. Next, slides were washed with 3× 5 min EBSS/sap and stained for α-SMA and Hoechst, as described above. Quantification of macrophages and arterial lumen area was performed as described above. CXCL10+ cells and IL-27+ cells were counted using ImageJ. Cell fluorescence was measured in the vessel wall with correction for background staining and expressed in arbitrary units (AUs).
2.4.3 Real-time PCR
Total RNA was isolated from adductor muscle according to the Trizol reagent protocol (Leiden University Medical Center, Leiden, The Netherlands). Total RNA was reverse-transcribed with the Revert Aid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). The resulting cDNA was amplified using 5 nmol/L of each primer and 50 nmol/L of Fast SYBR Green Master Mix (Invitrogen Life Technologies). Real-time PCR was performed as in the LDLR−/− protocol described above.
2.5 Statistical analysis
Results are presented as mean ± SEM. Significant differences between sample means were determined with an independent-sample t-test for data with a normal distribution and a non-parametric Mann–Whitney U-test for data with a non-normal distribution using GraphPad Prism 5 (GraphPad Software, Inc., CA, USA). LDPI data were statistically evaluated using a repeated-measures ANOVA test. A value of P < 0.05 was considered significant.
3. Results
3.1 Selection of antibody
Three different antibodies were tested for their capacities to inhibit the activity of IFNβ; two MAbs were directed against IFNβ and one against the IFNAR1 (a common receptor chain for IFNα and IFNβ). We stimulated mouse macrophages with murine INFβ, and measured the expression of IFNβ-response gene MX1 as readout for IFNβ activity (see Supplementary material online, Figure S1). All three antibodies effectively inhibited IFNβ activity. Because the IFNAR1 antibody showed the highest level of inhibition, we selected this antibody for subsequent use in our in vivo experiments.
3.2 Primary animal experiments in LDLR−/− mice
To analyse the effect of anti-IFNAR1 treatment on both arteriogenesis and atherosclerosis, we performed experiments in LDLR−/− mice that were subjected to femoral artery ligation. The study in LDLR−/− involved 4 weeks anti-IFNAR1 treatment in a setting of early atherosclerosis development. Three control animals died during the surgical procedure and these animals were excluded from the analysis.
3.2.1 LDPI in LDLR−/− mice
Hindlimb perfusion restoration was significantly improved in anti-IFNAR1-treated animals compared with controls at 7, 14, and 28 days (control vs. treatment; day 7: 23.6 ± 8.7 vs. 35.6 ± 16.5%, P = 0.03; day 14: 35.0 ± 12.3 vs. 51.4 ± 17.2%, P = 0.01; day 28: 54.0 ± 16.3 vs. 71.5 ± 13.8%, P = 0.01; ANOVA P = 0.004; Figure 1A and B). Notably, no signs of necrosis of the paws were detected in anti-IFNAR1- or control-treated mice and the anti-IFNAR1 showed the same weight gain as the control mice (data not shown).
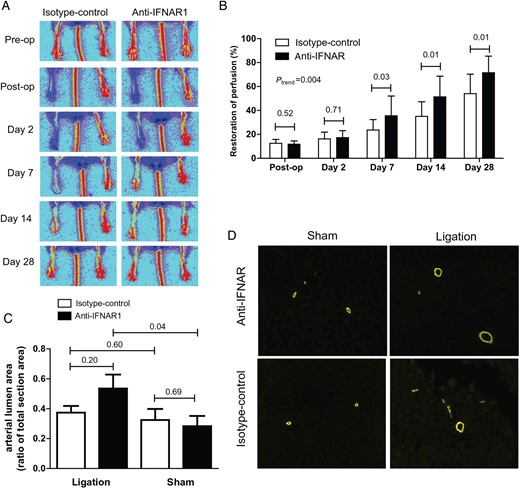
Arteriogenesis in 4-week-treated LDLR−/− mice. (A) Restoration of hindlimb perfusion in LDLR −/− mice after ligation of the femoral artery imaged by LDPI. (B) Four-week treatment of LDLR−/− mice with anti-IFNAR1 and follow-up with LDPI. Restoration of hindlimb perfusion was significantly improved in mice treated with anti-IFNAR1 compared with controls at 7, 14, and 28 days after femoral artery ligation. (C) Quantification of arterial lumen area as a ratio of total section area in hindlimb tissue by α-SMA staining. (D) Representative IF stainings (×10) for SMA from adductor muscle tissue. Shown are mean ± SEM. n = 13–16/group.
3.2.2 Histological analysis of arteriogenesis and angiogenesis
Quantification of arterial lumen area in adductor muscle tissue by α-SMA staining showed no difference in the arterial lumen area as a ratio of total section area after 4 weeks in treated LDLR−/− animals compared with controls (control vs. treatment: 0.37 ± 0.04 vs. 0.54 ± 0.09, P = 0.20). Arterial lumen area was significantly larger on the ligated side compared with the sham side in anti-IFNAR1-treated animals (0.54 ± 0.09 vs. 0.28 ± 0.07, P = 0.04; Figure 1C and D). Also, the number of arterioles per µm2 was similar (control vs. treatment: 3.79 × 10−6 ± 5.14 × 10−7 vs. 3.28 × 10−6 ± 4.54 × 10−7, P = 0.46; see Supplementary material online, Figure S2A and B). Capillary density expressed in the number of capillaries per mm2 was similar in treated animals compared with controls (control vs. treatment: 588.7 ± 23.0 vs. 632.7 ± 54.2, P = 0.50; see Supplementary material online, Figure 2C).
3.2.3 Atherosclerotic lesion analysis in LDLR−/− mice
Gene expression analyses of the aortic arches in the LDLR−/− mice demonstrated a decrease in IFNβ downstream target genes in the anti-IFNAR1-treated animals when compared with the controls following 4 weeks of treatment (STAT1 control vs. treatment: 1.00 ± 0.20 vs. 0.37 ± 0.04, P = 0.02; CXCL10: 1.00 ± 0.21 vs. 0.54 ± 0.11, P = 0.06; MX1: 1.00 ± 0.19 vs. 0.53 ± 0.15, P = 0.07; MX2: 1.00 ± 0.02 vs. 0.93 ± 0.01, P = 0.01; Figure 2A). No effects of anti-IFNAR1 therapy on gene expression in aortic arches of typical pro-inflammatory cytokines, such as TNF, IL-1β, and IL-6, could be observed (Figure 2B). Plasma cholesterol and triglyceride levels did also not differ between control and anti-IFNAR1-treated mice (see Supplementary material online, Figure S3A and B). In addition, no effect of anti-IFNAR1 treatment on relative leucocyte subsets was observed (see Supplementary material online, Figure S3C). Total atherosclerotic lesion area showed no difference between the controls and the treatment group after 4 weeks of treatment (Figure 3A and B). In addition, lesion distribution into early, moderate, or severe lesions did not show a difference in plaque severity between control and anti-IFNAR1-treated animals (Figure 3C). A more extensive immunohistochemical analysis of the aortic root demonstrated no differences in plaque phenotype, regarding collagen deposition, neutrophils, and T-cells as related to the lesion area (Figure 3D–G). Also, no difference was found in the amount of necrosis as a percentage of lesion area (Figure 3H). However, a significant decrease in apoptosis was observed in the anti-IFNAR1-treated mice (control vs. treatment: 0.16 ± 0.05 vs. 0.04 ± 0.01, P = 0.04; Figure 4A and B).
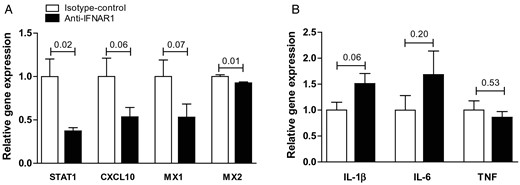
Systemic anti-IFNAR1 treatment blocks IFNβ signalling in vivo, but does not affect general inflammation in the aortic arch of LDLR−/− mice. (A) Relative gene expression of IFNβ downstream target genes in aortic arches of LDLR −/− mice treated with anti-IFNAR1 and controls in the 4-week experiments. (B) Relative aortic arch gene expression of pro-inflammatory cytokines. Shown are mean ± SEM. n = 13–16/group.
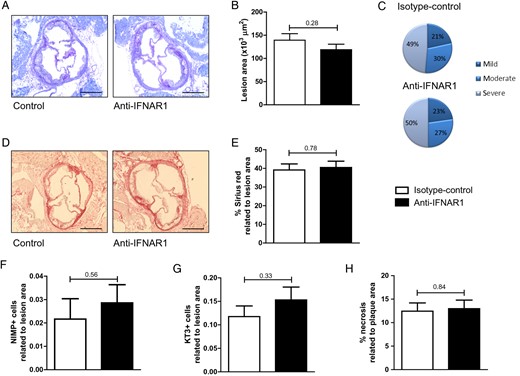
Systemic IFNAR1 blockade has no effect on atherosclerotic plaque size or phenotype. (A) Representative toluidin blue staining (×5) of aortic root tissue from controls and treated LDLR −/− mice. Scale bars represent 500 μm. (B) Quantification of lesion size in aortic root lesions. (C) Plaque severity score. (D) Representative Sirius Red-stained lesions (×5) from control and anti-IFNAR1-treated LDLR −/− mice. Scale bars represent 500 μm. (E) Quantification of collagen deposition in aortic root lesions. (F) Neutrophil and (G) T-cell counts in aortic root tissue from LDLR −/− mice following 4 weeks of control or anti-IFNAR1 treatment and (H) quantification of the necrotic core in the aortic root lesions. Shown are mean ± SEM. n = 13–16/group.
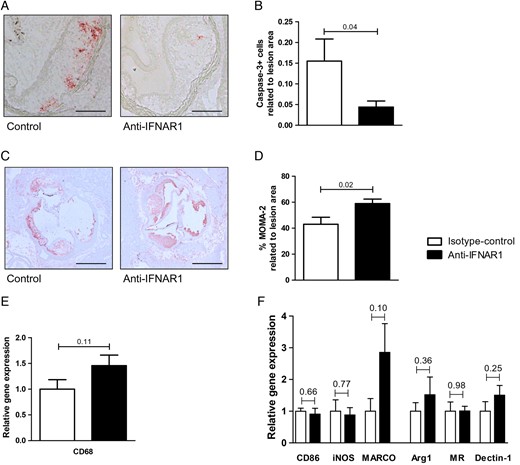
Systemic IFNAR1 blockade results in increased macrophage area and decreased apoptosis in the atherosclerotic plaque. (A) Representative caspase-3 staining in aortic root tissue from LDLR−/− mice following 4 weeks of control or anti-IFNAR1 treatment. Scale bars represent 500 μm. (B) Quantification of caspase-3-positive cells in aortic root lesions. (C) Representative MOMA-2 staining (×5) in aortic root tissue. Scale bars represent 500 μm. (D) Quantification of MOMA-2 in the aortic root lesions. (E) Relative gene expression of CD68 and (F) M1 and M2 macrophage markers in aortic arches of LDLR−/− mice. Shown are mean ± SEM. n = 13–16/group.
Further immunohistochemical analysis demonstrated a significant increase in the percentage of macrophage area in the lesions of anti-IFNAR1-treated animals (control vs. treatment: 43.0 ± 5.4 vs. 59.0 ± 3.5% of plaque area, P = 0.02; Figure 4C and D). In addition, a trend towards an increased gene expression of the macrophage marker CD68 was present in the aortic arches of anti-IFNAR1-treated animals (control vs. treatment: 1.0 ± 0.2 vs. 1.5 ± 0.2, P = 0.11, Figure 4E). This points to an increased macrophage accumulation in the plaques of those animals. However, gene expression of M1 and M2 macrophage markers did not differ between control and anti-IFNAR1-treated animals (Figure 4F).
3.3 Effect of treatment at early time point
3.3.1 Histological analysis of arteriogenesis, macrophages, and angiogenesis
To assess early changes upon ligation, we performed a short-term 2-day experiment. Quantification of arterial lumen area in calf tissue by α-SMA staining showed no difference in the arterial lumen area as a ratio of total section area after 2 days in treated LDLR−/− animals compared with controls (control vs. treatment: 0.09 ± 0.03 vs. 0.17 ± 0.08, P = 0.40; see Supplementary material online, Figure S4A). The number of arterioles per µm2 was similar in calf tissue 2 days following ligation in treated animals and controls (control vs. treatment: 3.65 × 10−6 ± 5.19 × 10−7 vs. 2.69 × 10−6 ± 6.20 × 10−7, P = 0.26). Fewer arterioles per µm2 were found on the ligated side when compared with the sham side in anti-IFNAR1-treated animals (2.70 ± 0.62 vs. 4.84 ± 0.72, P = 0.048) and controls (3.65 ± 0.52 vs. 5.44 ± 0.46, P = 0.03; see Supplementary material online, Figure S4B). Capillary density expressed in the number of capillaries per mm2 in calf tissue after 2 days was similar in treated animals compared with controls (control vs. treatment: 762.7 ± 158.9 vs. 638.0 ± 92.3, P = 0.52; see Supplementary material online, Figure S4C).
The mean number of surrounding macrophages per vessel in sections from hindlimb tissue from the LDLR−/− mice did not differ between mice treated with anti-IFNAR1 and controls (control vs. treatment: 5.3 ± 0.3 vs. 5.1 ± 0.3, P = 0.62). Furthermore, the mean number of vessels with surrounding macrophages per fixed section area did not differ significantly (control vs. treatment: 1.3 ± 0.04 vs. 1.3 ± 0.05, P = 0.96). We found no differences in M1 and M2 infiltration in ligated adductor muscle tissue between anti-IFNAR1-treated animals and controls [ratio of CD40-positive cells (M1) vs. F4/80-positive cells (all macrophages); control vs. treatment: 0.81 ± 0.03 vs. 0.77 ± 0.03, P = 0.23; ratio of MR-positive (M2) vs. F4/80-positive cells (all macrophages); control vs. treatment: 0.08 ± 0.01 vs. 0.07 ± 0.01, P = 0.52]. Also, we found no significant differences in the mean number of surrounding neutrophils per vessel between anti-IFNAR1-treated animals and controls (control vs. treatment: 2.59 ± 0.20 vs. 2.50 ± 0.14, P = 0.71; see Supplementary material online, Figure S5A). However, a significant higher mean number of surrounding T-cells per vessel was found in animals treated with IFNAR1 antibodies compared with controls (control vs. treatment: 1.50 ± 0.11 vs. 2.85 ± 0.22, P < 0.0001; see Supplementary material online, Figure S5B).
3.3.2 Systemic inflammation
Anti-IFNAR1 treatment had no effect on systemic inflammation after 2 days of treatment, as the percentage of leucocyte subsets and pro-inflammatory M1 markers on blood monocytes were not different between control and treated mice (see Supplementary material online, Figure S6).
3.4 Animal experiments in APOE−/− mice
The study in APOE−/− mice was performed to investigate the effect of anti-IFNAR1 treatment on arteriogenesis and advanced atherosclerosis with a short 1-week anti-IFNAR1 intervention. Two animals receiving treatment died and these animals were excluded from the analysis.
3.4.1 Laser Doppler perfusion imaging
In accordance with the results obtained from the 4-week-treated LDLR−/− mice experiments, hindlimb perfusion restoration was also significantly improved in APOE−/− mice treated with anti-IFNAR1 compared with controls at 2 and 7 days after femoral artery ligation (control vs. treatment; day 2: 18.8 ± 4.2 vs. 27.6 ± 9.7%, P = 0.01; day 7: 31.1 ± 5.9 vs. 45.9 ± 6.2%, P = 0.04; ANOVA, P = 0.015; Figure 5A).
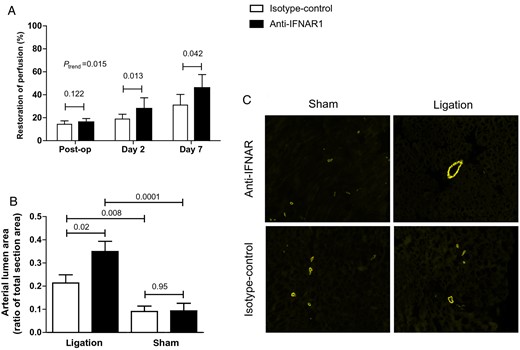
Arteriogenesis in 1-week-treated APOE−/− mice. (A) One-week treatment of APOE−/− mice with anti-IFNAR1 and follow-up with LDPI. Restoration of hindlimb perfusion was significantly improved in mice treated with anti-IFNAR1 compared with controls at 2 and 7 days after femoral artery ligation. (B) Quantification of arterial lumen area as a ratio of total section area in hindlimb tissue by α-SMA staining. (C) Representative IF stainings (×10) for SMA from adductor muscle tissue. Shown are mean ± SEM. n = 14–12/group.
3.4.2 Histological analysis of arteriogenesis, macrophages, and angiogenesis
Quantification of arterial lumen area as a ratio of total section area in hindlimb tissue by α-SMA staining also showed an increased arterial lumen area after 1 week in treated APOE−/− animals compared with controls (control vs. treatment: 0.21 ± 0.04 vs. 0.35 ± 0.04, P = 0.02). Arterial lumen area was also significantly larger on the ligated side compared with the sham side in anti-IFNAR1-treated animals (0.35 ± 0.04 vs. 0.09 ± 0.03, P = 0.0001) and in control animals (0.22 ± 0.04 vs. 0.09 ± 0.02, P = 0.008; Figure 5B and C).
The mean number of surrounding macrophages per vessel in sections from hindlimb tissue from the APOE−/− mice did not differ between mice treated with anti-IFNAR1 and controls (control vs. treatment: 3.9 ± 0.6 vs. 4.0 ± 0.4, P = 0.26). Furthermore, the mean number of vessels with surrounding macrophages per fixed section area did not differ significantly (control vs. treatment: 6.5 ± 0.9 vs. 6.8 ± 1.0, P = 0.62). No difference was found in the distribution of macrophage subsets surrounding the vessels [ratio of CD40-positive cells (M1) vs. F4/80-positive cells (all macrophages); control vs. treatment: 0.05 ± 0.03 vs. 0.08 ± 0.02, P = 0.13]. In both groups, very few M1 macrophages were seen. The ratio of MR-positive (M2) vs. F4/80-positive cells (all macrophages) was also similar in both groups (control vs. treatment: 0.77 ± 0.05 vs. 0.75 ± 0.03, P = 0.92).
The number of arterioles per µm2 was similar (control vs. treatment: 2.65 ± 0.39 vs. 2.87 ± 0.36, P = 0.68) (see Supplementary material online, Figure S7A). Fewer arterioles per µm2 were found on the ligated side when compared with the sham side in anti-IFNAR1-treated animals (1.41 ± 0.25 vs. 2.87 ± 0.36, P = 0.003) and controls (0.91 ± 0.11 vs. 2.65 ± 0.39, P = 0.0001). Also, capillary density expressed in the number of capillaries per mm2 was similar in treated animals compared with controls (control vs. treatment: 747.1 ± 105.3 vs. 678.1 ± 57.3, P = 0.59; see Supplementary material online, Figure S7B).
3.4.3 Gene expression analysis arteriogenesis
In search of mechanistic evidence responsible for increased arteriogenesis, we performed real-time RT-PCR for major factors known to influence arteriogenesis in ischaemic hindlimb tissue of anti-IFNAR1-treated mice and controls. PDGF-a and FGF-2 showed similar expression, whereas VEGF-a showed lower expression in hindlimb tissue from anti-IFNAR1-treated animals compared with controls (control vs. treatment: 1.84 ± 0.50 vs. 0.63 ± 0.15, P = 0.03; see Supplementary material online, Figure S8).
3.4.4 IFNβ downstream targets: ligated vs. sham hindlimb
Cell fluorescence of two IFNβ downstream targets, CXCL10 and IL-27, was compared between sham (left) hindlimb and ligated (right) hindlimb. At day 7, a significantly higher intensity of CXCL10 in the vessel wall was found at the ligated side in comparison with the sham side while a trend was found for IL-27 (CXCL10, sham vs. ligated: 458.7 ± 68.2 vs. 951.1 ± 60.4, P < 0.001; IL-27, sham vs. ligated: 253.2 ± 64.1 vs. 395.3 ± 37.7, P = 0.05). Also, the number of CXCL10- and IL-27-positive cells was higher in hindlimb tissue from the ligated side compared with the sham side (CXCL10, sham vs. ligated: 6.9 ± 0.8 vs. 10.2 ± 0.4, P < 0.001; IL-27, sham vs. ligated: 1.9 ± 0.4 vs. 5.3 ± 0.5, P < 0.001). Real-time RT-PCR analysis correspondingly showed increased expression of CXCL10 in ligated compared with sham adductor muscle (sham vs. ligated: 0.03 ± 0.01 vs. 1.1 ± 0.4, P = 0.02; Figure 6A and B). These data confirm induction of an IFN response upon femoral ligation.
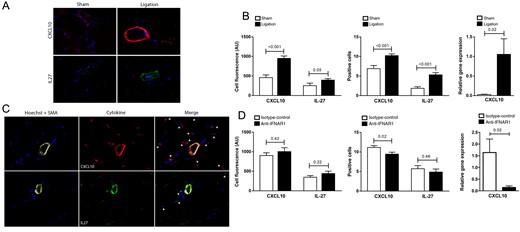
IFNβ downstream targets in 1-week-treated APOE−/− mice. (A) Representative IF stainings (×40) for CXCL10 and IL-27 from sham (left) and ligated (right) adductor muscle tissue from the hindlimb of APOE−/− mice. (B) Cell fluorescence of CXCL10 and IL-27 in the vessel wall from sections of the sham and ligated side in AUs, positive cell count of CXCL10 and IL-27 in hindlimb tissue from the sham and ligated side, and real-time RT-PCR analysis of relative gene expression of CXCL10 in hindlimb tissue from the sham and ligated side in AUs. (C) Representative IF stainings (×40) for CXCL10, IL-27, and α-SMA from adductor muscle tissue from the hindlimb of a control mouse. (D) Cell fluorescence of CXCL10 and IL-27 in the vessel wall from sections of treated mice and controls in AUs, positive cell count of CXCL10 and IL-27 in hindlimb tissue from treated mice and controls, and real-time RT-PCR analysis of relative gene expression of CXCL10 in hindlimb tissue from treated mice and controls in AUs. Shown are mean ± SEM. n = 14–12/group.
3.4.5 IFNβ downstream targets: anti-IFNAR1-treated mice vs. control
When comparing anti-IFNAR1-treated mice with control, no significant differences were found in total cell fluorescence of CXCL10 and IL-27 in the vessel wall (CXCL10, control vs. treatment: 902.6 ± 72.0 vs. 1004 ± 99.9, P = 0.42; IL-27, control vs. treatment: 348.3 ± 42.6 vs. 437.2 ± 61.5, P = 0.22). However, upon quantification of cells, a decreased number of CXCL10-positive cells was found in hindlimb tissue from the anti-IFNAR1-treated group (control vs. treatment: 11.1 ± 0.5 vs. 9.5 ± 0.5, P = 0.02). IL-27-positive cell count was not significantly different between the groups (control vs. treatment: 5.7 ± 0.8 vs. 4.9 ± 0.8, P = 0.46). These findings were corroborated by significant higher levels of CXCL10 gene expression in control mice compared with anti-IFNAR1-treated mice (control vs. treatment: 1.6 ± 0.6 vs. 0.1 ± 0.1, P = 0.02; Figure 6C and D).
3.4.6 Atherosclerosis assessment
Atherosclerotic lesion analyses were also in line with the 4-week experiments showing no significant differences between the anti-IFNAR1 and control group when examining general plaque characteristics and severity (Figure 7A and B). In addition, no effects of anti-IFNAR1 therapy on gene expression of typical pro-inflammatory cytokines, such as TNF, IL-1β, and IL-6, in aortic arches could be observed (Figure 7C).
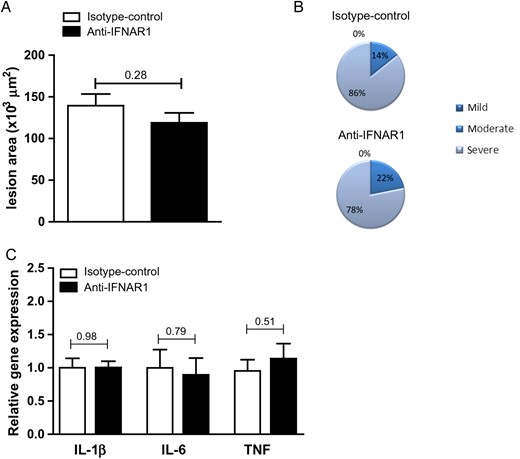
Systemic IFNAR1 blockade does not affect atherosclerotic lesion size and does not affect pro-inflammatory cytokines in the aortic arch of 1-week-treated APOE−/− mice. (A) Quantification of lesion size and (B) plaque severity score in aortic root lesions of APOE−/− mice. (C) Gene expression in aortic arches of APOE−/− mice following 1 week of anti-IFNAR1 treatment. Shown are mean ± SEM. n = 14–12/group.
4. Discussion
In this study, we investigated the effect of blocking IFNAR1 on arteriogenesis and atherosclerosis using in vivo mouse models. For the first time, we show that mAb therapy against IFNAR1 leads to improved restoration of hindlimb perfusion after femoral artery ligation in mice, without enhancing atherosclerotic burden. Application of mAb therapy has proved to be an efficacious treatment in multiple diseases, like cancer and rheumatic diseases.16,17 In the field of cardiovascular disease, MAbs are currently being investigated, for example to reduce low-density lipoprotein cholesterol levels by blocking PCSK918 or to reduce the inflammatory profile by blocking IL-1β.19 MAbs were also previously tested to stimulate arteriogenesis. Hellingman et al.20 expanded regulatory T-cell numbers two-fold in a murine ischaemic hindlimb model by injecting mice with IL-2 mixed with an IL-2 MAb. They showed a significant attenuation of blood flow recovery on LDPI and also a significant lower number of α-SMA-expressing collaterals in the hindlimb tissue on the ligated side in animals treated with antibody compared with controls.20 Depletion of regulatory T-cells with anti-CD25 treatment, however, showed no effect on arteriogenesis in this study.
Mouse MAbs specific for the IFNAR1 subunit of the mouse IFN-α and -β receptor were previously generated in IFNAR1−/− mice. Clone MAR1-5A3 was shown to block type I IFN-induced antiviral, antimicrobial, and antitumor responses in vivo.11 Increased sensitivity to viral infection was shown to be similar in IFNAR1 knockout mice when compared with antibody-treated wild-type mice.21 The MAR1-5A3 clone was also shown to delay CD8+ T-cell maturation during viral infection.22 In accordance, our MX-1 assay showed a strong blocking capacity of MAR1-5A3; therefore, we selected this clone for application in our in vivo experiments.
In previous studies, we found IFNβ expression to be up-regulated in monocytes isolated from patients with a poor arteriogenic response.6 Furthermore, arteriogenesis is hampered after exogenous application of IFNβ in a murine hindlimb model.6 IFNAR1−/− mice showed improved hindlimb perfusion 1 week after femoral artery ligation compared with wild-type mice as assessed by infusion of fluorescent microspheres.7 In the present study, we show that the inhibitory effect of IFNβ on arteriogenesis can be inverted by blocking IFNAR1. Hindlimb perfusion restoration after femoral artery ligation was improved in mice treated with anti-IFNAR1 compared with controls as assessed by LDPI. An increased arterial lumen area in animals treated for 1 week endorses the finding of an accelerated growth of collateral arteries. The lack of a significant difference in arterial lumen area in animals treated for 2 days and 4 weeks may be attributed to the fact that perfusion depends on luminal size to the fourth power and consequently, relatively small changes in vessel diameter can result in large changes in perfusion, as seen by LDPI. Furthermore, we show that in hindlimb tissue, the IFNAR1 blockade leads to a decreased number of CXCL10-positive cells and a lower CXCL10 gene expression and likewise to a decrease in CXCL10 gene expression in the aortic arches. The mechanism through which CXCL10 mediates its effects on arteriogenesis is not completely elucidated. van den Borne et al.23 investigated the role of CXCL10 in arteriogenesis and showed that bone marrow-derived CXCL10 and tissue-derived CXCL10 play a role in accelerating perfusion recovery after arterial occlusion in mice. They postulate that this is probably induced by the promotion of vascular smooth muscle cell recruitment and maturation of pre-existing anastomoses. The discrepancy between our results and those of van den Borne et al. can be explained by the fact that CXCL10 has both pro-arteriogenic23 as well as anti-arteriogenic effects.24,25,26,27 van den Borne et al. demonstrated that in the early phase after femoral artery ligation, CXCL10 expression in hindlimb tissue increases, with improved restoration of perfusion. In the late phase, however, CXCL10 expression subsides and the rapid perfusion recovery as seen in the early phase is diminished. This anti-arteriogenic effect of CXCL10 in the late phase corresponds with our findings that restoration of perfusion is accelerated in the late phase following ligation accompanied by lowered levels of CXCL10. Moreover, it is important to take into account that CXCL10−/− mice possibly have an altered immune system causing differences in endothelial cell proliferation, making comparison with previous studies as well as our own work challenging.
Studies investigating the role of macrophage subpopulations in arteriogenesis have shown that a shift towards M2 macrophages improves collateral remodelling.28 However, we found that no difference in the distribution of macrophage subsets between the groups and the total number of vessels with surrounding macrophages was similar.
In search of mechanistic evidence responsible for increased arteriogenesis, we performed PCR for major factors known to influence arteriogenesis in ischaemic hindlimb tissue of anti-IFNAR1-treated mice and controls. PDGF-a and FGF-2 showed similar expression, whereas VEGF-a showed lower expression in hindlimb tissue from anti-IFNAR1-treated animals compared with controls. VEGF-a is known to induce a heterogeneous response29 and its inducing effect on angiogenesis is more distinct than that of arteriogenesis.30 Our results cannot confirm a role for aforementioned growth factors in promoting collateral vessel growth. Thacker et al.31 showed that IFNAR1 knockout mice show enhanced endothelial-dependent vasodilation. Possibly, the IFNAR1 blockade as performed in our experiments leads to enhanced blood flow recovery through this mechanism. Another possible mechanism behind the enhanced collateralization found in our experiments could be T-cell-dependent vascularization. Stabile et al.32 showed that CD4+ T-cells control the arteriogenic response to acute hindlimb ischaemia. Sharir et al.33 found that regulatory T-cells play an important role in flow re-establishment after inducement of hindlimb ischaemia. In line with these studies, we found significantly elevated numbers of CD3+ T-cells in hindlimb tissue from anti-IFNAR1-treated animals compared with controls.
Increased atherosclerotic plaque formation and plaque destabilization have hindered the development of therapeutic pro-arteriogenic therapies.10 For example, MCP-1 was shown to be a potent pro-arteriogenic chemokine.34 However, local application caused systemic monocyte activation and effects on neointima and plaque composition leading to potentially unstable atherosclerotic lesions,10 rendering its pharmaceutical application unsuitable for the treatment of CAD.
Although type I IFNs have important immunomodulatory functions, primarily in response to viral encounters,35,36 they also have been linked to disease progression, as they are for instance implicated in the pathogenesis of SLE.37 Interestingly, patients with SLE show an increased risk of cardiovascular disease, which is directly associated with their increased type I IFN levels.38 The involvement of type I IFNs in atherosclerosis was further illustrated by the observation that the main cellular source of type I IFN, plasmacytoid dendritic cells (pDCs), is present in rupture-prone areas of the human atherosclerotic plaque.39 Furthermore, pDC depletion in APOE−/− mice decreased lesion size, and similar reductions in lesion size were obtained in LDLR−/− mice with MHC class II-deficient pDCs, which indicates that pDCs promote atherosclerosis.40,41 In addition, we recently demonstrated that vulnerable plaques have increased type I IFN signalling as well.9 Thus, type I IFNs are associated with plaque instability in human lesions.
In our experiments, we examined the effect of systemic IFNAR1 blockade on plaque size and composition using a well-established mouse model of atherosclerosis, the LDLR−/− mouse.42 Using this model, we observed no effects of anti-IFNAR1 treatment on total plaque size following treatment. An enhanced macrophage accumulation was found; however, M1 and M2 macrophage markers in the aortic arch did not differ between control and treated animals. Thus, even though an increase in macrophages may be present, these macrophages do not display an increased pro-inflammatory phenotype. Furthermore, relative blood leucocyte levels were similar between control and anti-IFNAR1-treated animals, which we could confirm in the 2-day experiments, indicating that anti-IFNAR1 treatment has no effect on systemic inflammation either. The increased macrophage accumulation was also accompanied by decreased apoptosis in the lesions of anti-IFNAR1-treated animals, indicating improved survival of foam cells upon blockade of IFN signalling. We previously demonstrated that absence of myeloid IFNAR1 signalling is also protective for foam cell death9 and we can thus recapitulate this with a systemic antibody blockade. Such decrease in apoptosis may be considered beneficial, as the resulting decrease in cell debris limits necrotic core formation, which strongly affects plaque vulnerability.43 This is in line with our previous results, showing pro-apoptotic effects of IFNβ via increased expression of TRAIL, FAS, and FASL, as well as decreased apoptosis in monocytes derived from IFNAR1−/− mice.7 Of note, IFNβ is also known to prime monocyte-derived macrophages for cell death in multiple sclerosis (MS), in which a subgroup of patients is treated with IFNβ.44
In a previous study,9 we observed a reduction in atherosclerosis development, with less macrophage accumulation and a decrease in plaque cell death, by the genetic myeloid-specific ablation of IFNAR1. In the present study, we did not observe such reduction in atherosclerosis. Possibly, the anti-IFNAR1 treatment period in the present study was too short. In addition, the systemic antibody-mediated approach, as studied in the present manuscript, also targets the endothelium which might counteract the myeloid effects observed previously.9 We indeed observed a significantly increased expression of the endothelial cell activation marker VCAM1 in lesions of anti-IFNAR1-treated animals, with a concomitant increase in VCAM1 aortic gene expression (data not shown), which demonstrates that in atherosclerosis, type I IFNs might prevent endothelial activation. Interestingly, part of the beneficial effects of IFNβ treatment in MS patients is attributed to an indirect blockade of cerebral endothelial activation. IFNβ treatment increases serum levels of soluble VCAM1, which binds to its co-receptor VLA-4 on leucocytes. This subsequently inhibits leucocyte adhesion to membrane-bound VCAM1, resulting in less cellular recruitment to inflammatory foci and dampening clinical relapses.45
A similar counteracting factor might be the targeting of T regulatory cells (Tregs). Tregs are atheroprotective as depletion of CD4+CD25+ Tregs, aggravated atherosclerosis in LDLR−/− mice, whereas adoptive transfer of CD4+CD25+ Tregs diminished atherosclerosis in APOE−/− mice.46 In the spleen of the anti-IFNAR1-treated LDLR−/− mice, we observed less suppressive Tregs when compared with control-treated mice, as the gene expression of FoxP3 and TGF-β, its key cytokine, decreased (data not shown). This further demonstrates the importance of cell-specific targeting, making the myeloid compartment an attractive target for further studies, where we will specifically focus on myeloid IFNAR1 signalling to stimulate arteriogenesis with an additional reduction in atherosclerosis development and without side effects on endothelium or T-cell populations.
5. Conclusion
The findings of the present study indicate that blocking IFNAR1, using MAbs during both a 1- and 4-week period, stimulates collateral artery growth in mice without enhancing atherosclerotic burden. This is the first successful approach to stimulate arteriogenesis using MAbs, paving the way towards clinical application of such strategy.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by ICAR-VU (2010, ‘Regenerative therapies to treat ischemic heart disease’ to N.v.R. and A.J.H.) and the Netherlands Heart Foundation (grant 2010B022 to M.P.d.W.). M.P.d.W. is an established investigator of the Netherlands Heart Foundation (2007T067). We also acknowledge the support from the Netherlands CardioVascular Research Initiative, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research, and Development and the Royal Netherlands Academy of Sciences for the GENIUS project ‘Generating the best evidence-based pharmaceutical targets for atherosclerosis' (CVON2011-19).
Acknowledgements
We thank the staff members of the animal laboratory facility and Anouk Haverkamp, Ryanne Betgem, Guus de Waard, Sylvia Nieuwenhuis, and Josefien Baggen for skilled assistance during the various procedures.
Conflict of interest: none declared.
References
Author notes
P.F.A.T., M.C.B., M.P.d.W., and N.v.R. contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}