





Abstract
Both mitochondria and nitric oxide (NO•) contribute to cardioprotection by ischaemic preconditioning (IPC). IPC causes mild uncoupling of mitochondria via uncoupling proteins (UCPs) and the adenine nucleotide translocase (ANT), and mild uncoupling per se is cardioprotective. Although electrophilic lipids are known to activate mitochondrial uncoupling, the role of such species in IPC-induced uncoupling and cardioprotection is unclear. We hypothesized that endogenous formation of NO•-derived electrophilic lipids (nitroalkenes such as nitro-linoleate, LNO2) during IPC may stimulate mitochondrial uncoupling via post-translational modification of UCPs and ANT, thus affording cardioprotection.
Hearts from male Sprague-Dawley rats were Langendorff-perfused and subjected to IPC. Nitroalkene formation was measured by HPLC-ESI-MS/MS. The effects of exogenous LNO2 and biotin-tagged LNO2 on isolated heart mitochondria and cardiomyocytes were also investigated.
Nitroalkenes including LNO2 were endogenously generated in mitochondria of IPC hearts. Synthetic LNO2 (<1 µM) activated mild uncoupling, an effect blocked by UCP and ANT inhibitors. LNO2 (<1 µM) also protected cardiomyocytes against simulated ischaemia–reperfusion injury. Biotinylated LNO2 covalently modified ANT thiols and possibly UCP-2. No effects of LNO2 were attributable to NO• release, cGMP signalling, mitochondrial KATP channels, or protective kinase signalling.
Components of a novel signalling pathway are inferred, wherein nitroalkenes formed by IPC-stimulated nitration reactions may induce mild mitochondrial uncoupling via post-translational modification of ANT and UCP-2, subsequently conferring resistance to ischaemia–reperfusion injury.
1. Introduction
Cardiac ischaemic preconditioning (IPC) is an endogenous protective mechanism, in which short cycles of non-lethal ischaemia–reperfusion (IR) elicit protection from subsequent prolonged IR injury.1 The mechanisms underlying IPC-mediated cardioprotection are debated, but a consensus has emerged that nitric oxide (NO•) and mitochondria play essential roles.2–6 Despite this consensus, links between NO• signalling and effector mechanisms at the mitochondrial level remain elusive. For example, while NO• signalling via cGMP-dependent protein kinase (PKG) can phosphorylate several mitochondrial targets of relevance to IPC,7–9 the importance of PKG-independent effects of NO• on mitochondria is less clear.4 Similarly, mild uncoupling of mitochondria is an important IPC-induced protective event,10,11 but its potential upstream regulation by NO• is unclear. The aim of this study was to elucidate novel mechanisms linking NO• and mitochondria in IPC.
One unexplored aspect of NO• signalling in IPC is the nitration of unsaturated fatty acids to yield electrophilic nitroalkene derivatives (e.g. nitro-linoleate and nitro-oleate, LNO2 and OA-NO2, respectively).12 Although the biochemical mechanisms of lipid nitration are not fully elucidated,13 nitroalkenes are found endogenously in humans14 and can mediate pluripotent cell signalling effects.15 These effects may be mediated by electrophilic reaction of nitroalkenes with protein thiols, to form covalent ‘nitroalkylation’ adducts.16 Notably, conditions during IPC could favour nitroalkene generation from the abundance of polyunsaturated fatty acids in mitochondrial membranes.17 These conditions include elevated NO•, 5,6 transient reactive oxygen species (ROS) generation,18,19 acidic pH,13 and the activation of both lipoxygenases and mitochondrial phospholipase A2.20,21 In addition, both peroxynitrite and electrophilic lipids can activate mitochondrial uncoupling,22,23 and mild uncoupling itself is cardioprotective.11,24 Combining these observations we hypothesized that nitroalkenes may be formed in mitochondria during IPC, and may nitroalkylate mitochondrial proteins thereby activating mild uncoupling, leading to cardioprotection against IR injury.
2. Methods
Detailed experimental procedures are given in the Supplementary material online. All chemicals were of the highest grade available from Sigma (St Louis, MO, USA) unless otherwise stated. LNO2 and biotinylated LNO2 (Bt-LNO2) were synthesized, purified, quantified, and stored as previously,14,25,26 with all procedures performed under subdued light. Non-nitrated linoleic acid (LA) served as a control throughout.
Male Sprague-Dawley rats (Harlan, Indianapolis, IN, USA), 200–250 g body mass, were housed in accordance with the NIH Guide for the Care and Use of Laboratory Animals (US National Institutes of Health Publication No. 85–23, revised 1996). All procedures were also approved by the University of Rochester Committee on Animal Resources (UCAR, protocols 2003-111 and 2007-087). Hearts were perfused as previously,10 and subjected to either (i) normoxic perfusion, (ii) IPC, (iii) IPC plus the NO• synthase inhibitor l-nitro-arginine methyl ester (l-NAME, 100 µM), (iv) ischaemia, or (v) IPC plus ischaemia.
Heart mitochondria were isolated and protein determined as previously.10 Mitochondrial respiration and uncoupling were measured as previously.10 Optional additions to incubations were: 1–5 µM LNO2, 1 mM guanosine diphosphate (GDP), or 50 µM Genipin (Wako, Richmond, VA, USA)27 to inhibit uncoupling proteins (UCPs) or UCP-2, respectively; 5 µM carboxyatractyloside (CATr, Calbiochem, San Diego, CA, USA) to inhibit adenine nucleotide translocase (ANT); 20 µM ethanethiol (E-SH) to reverse thiol modifications; 30 µM 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide (c-PTIO, Axxora, San Diego, CA, USA) to scavenge NO•; or 1 mM 2,6-di-tert-butyl-4-methyl-phenol (BHT), a lipid-soluble antioxidant. Mitochondrial permeability transition (PT) pore opening was measured as previously.28
Adult rat ventricular cardiomyocytes were isolated and state 4 respiration plus mitochondrial membrane potential (Δψm) were measured as previously.29 Cells were subjected to simulated ischaemia–reperfusion (SIR) injury as previously,29 with cell viability measured by Trypan blue exclusion. This model eliminates the potentially confounding vascular effects of LNO2.15 Briefly, SIR comprised 1 h hypoxia in glucose-free buffer at pH 6.5, then 30 min reoxygenation in glucose-replete buffer at pH 7.4. The following were optionally present: LNO2 (0.25–1 µM), LA (0.5–1 µM), the soluble guanylate cyclase (sGC) inhibitor 1H-[1,2,4]oxadiazole-[4,3-a]quinoxalin-1-one (ODQ, 10 µM), the mitochondrial KATP channel antagonists 5-hydroxydecanoate (5-HD, 300 µM) or glybenclamide (2 µM), the extracellular signal regulated kinase (ERK) inhibitor UO126 (10 µM), or the phosphoinositide 3 kinase (PI3K) inhibitor wortmannin (100 nM).
Biotinylated proteins were immunoprecipitated from Bt-LNO2-treated mitochondria or cardiomyocytes using neutravidin-agarose as previously.10 Immunoprecipitated samples or whole extracts were western blotted as previously,10 using antibodies against biotin, ANT, or UCP-2.
Lipids were extracted from 5 mg mitochondrial protein as previously.30 A synthetic [13C18]-LNO2 internal standard was added to correct for extraction losses. Nitroalkenes were detected by HPLC electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS) as previously.14 Biologically derived nitroalkenes were identified by retention time, precursor ion mass, MS/MS fragmentation pattern, and thiol reactivity,14 and were quantified relative to the internal standard.
All experiments were performed three to eight times, each n representing an independent mitochondrial, cell, or heart preparation (separate animal). Significance between groups was established by ANOVA.
3. Results
In the current study, we investigated whether fatty acid nitration could occur in cardiac mitochondria during IPC. Mitochondrial lipid extracts from perfused hearts were analysed by HPLC-ESI-MS/MS using a multiple reaction monitoring (MRM) transition of 324/46 in negative ion mode. The chromatograms (Figure 1A) revealed that mitochondria from IPC hearts contained elevated levels of an LNO2 nitroalkene derivative, with positional isomers eluting at identical times to a synthetic LNO2 nitroalkene standard. Concomitant product ion analysis (Figure 1B) revealed a fragmentation pattern for the IPC-derived nitroalkene consistent with the previously reported LNO2 structure.14 Quantitation of LNO2 via internal standard revealed its concentration in IPC mitochondria to be 619 ± 137 fmol/mg mitochondrial protein (Figure 1C). The NOS inhibitor l-NAME attenuated the IPC-induced increase in mitochondrial LNO2 by ∼60% (P = 0.06 vs. IPC), although it is not known if the l-NAME insensitive fraction of LNO2 is due to incomplete NOS inhibition or represents LNO2 generation from other reactive nitrogen species (RNSs) such as NO2−. Ischaemia alone generated a miniscule amount of LNO2, but notably in mitochondria from hearts subjected to IPC plus ischaemia, LNO2 levels dropped to ∼16% of those seen in IPC alone, suggesting rapid LNO2 degradation. Mitochondria also contained OA-NO2, but its levels did not change in IPC (215 ± 74 vs. 245 ± 41 fmol/mg protein in control vs. IPC, respectively). In addition nitroalkenes were detected in other subcellular compartments (data not shown). Due to space restrictions the current study focuses on mitochondrial IPC samples, and a more complete characterization of cardiac nitroalkenes during IPC and IR, including their metabolism by mitochondrial β-oxidation, is anticipated to be the subject of a subsequent manuscript.
![Endogenous LNO2 formation in mitochondria during IPC. (A) Lipid extracts were prepared from mitochondria isolated from control and IPC-treated hearts, and analysed by HPLC ESI-MS/MS in MRM mode using m/z 324/46 transition to identify LNO2. Blank solvent extract and synthetic standards were analysed by the same methods. Insets to chromatograms highlight the co-elution of LNO2 derived from IPC mitochondria with the synthetic LNO2 standard. Data are representative of n = 8 samples. (B) Product ion analysis of LNO2 derived from IPC-treated heart mitochondria shows the major fragment ions generated after collision-induced dissociation. Fragments at m/z 324, 306, 293, 288, and 277 are [M–H]−, [M–H2O]−, [M–HNO]−, [M–2H2O]−, and [M–HNO2]−, respectively. The major product ion, m/z 46 is the ionized nitro group (NO2−). The fragmentation pattern of IPC mitochondria-derived LNO2 is the same as that generated from synthetic LNO2 25. (C) Quantitation of LNO2 in mitochondrial samples, achieved by spiking mitochondria prior to lipid extraction with 500 pg of [13C18]LNO2 as internal standard (m/z 342). The relative peak areas of [13C18]LNO2 vs. endogenous LNO2 were used to quantify LNO2 in the original mitochondrial samples using an internal standard curve, and data were normalized to amount of mitochondrial protein. Data are means ± SEM, n = 4. *P < 0.05 vs. control. #P < 0.05 vs. IPC alone. Treatment groups are detailed in the methods.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/82/2/10.1093_cvr_cvn323/1/m_cvn32301.gif?Expires=1716515513&Signature=t8ut5W1MTjKQikolmEUJGlBVOUHKfYEvtgRjPHKvHYxXCv-Esz5QXdtZm7Nsza5CHLt7YNQP-cruRW90GQBddpcyGYnAIuwW3tFpj4RXRMHcnjqXMDZuv~tzuaLM6pJdXo2PJwAmG0kUPOCTy5UkU8mim9ctE-8kU28AGdHrrAJkLuTUA7FcxZj4PEAhtecBiOAcfODbfBPBTd-6R~YTwSTgLiFcinaSCy5axjX2MbhMB4mAJEgbHVlAt3fzCuvxLsj5Rgl-9ISojZoBa~NYICIBDeN-evIMCaAoVq9eyAgfP4KhTabUJrLHR9b48317BBeIxuAn5RXIuCjTmtPRqg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Endogenous LNO2 formation in mitochondria during IPC. (A) Lipid extracts were prepared from mitochondria isolated from control and IPC-treated hearts, and analysed by HPLC ESI-MS/MS in MRM mode using m/z 324/46 transition to identify LNO2. Blank solvent extract and synthetic standards were analysed by the same methods. Insets to chromatograms highlight the co-elution of LNO2 derived from IPC mitochondria with the synthetic LNO2 standard. Data are representative of n = 8 samples. (B) Product ion analysis of LNO2 derived from IPC-treated heart mitochondria shows the major fragment ions generated after collision-induced dissociation. Fragments at m/z 324, 306, 293, 288, and 277 are [M–H]−, [M–H2O]−, [M–HNO]−, [M–2H2O]−, and [M–HNO2]−, respectively. The major product ion, m/z 46 is the ionized nitro group (NO2−). The fragmentation pattern of IPC mitochondria-derived LNO2 is the same as that generated from synthetic LNO2 25. (C) Quantitation of LNO2 in mitochondrial samples, achieved by spiking mitochondria prior to lipid extraction with 500 pg of [13C18]LNO2 as internal standard (m/z 342). The relative peak areas of [13C18]LNO2 vs. endogenous LNO2 were used to quantify LNO2 in the original mitochondrial samples using an internal standard curve, and data were normalized to amount of mitochondrial protein. Data are means ± SEM, n = 4. *P < 0.05 vs. control. #P < 0.05 vs. IPC alone. Treatment groups are detailed in the methods.
Next, the potential for exogenous LNO2 to protect against SIR injury was tested in isolated cardiomyocytes. Figure 2A shows that LNO2 significantly improved post-SIR cardiomyocyte viability, with maximal protection at 0.5 µM LNO2. Non-nitrated LA was without effect, and the mito-KATP channel antagonists 5-HD or glybenclamide31 did not reverse the effect of LNO2, suggesting no role for this channel in LNO2-mediated protection. Notably in this system, 5-HD did block protection by the mito-KATP channel agonist diazoxide, indicating appropriate 5-HD efficacy (not shown). In addition, the sGC inhibitor ODQ,4–7 the ERK inhibitor UO-126,32 and the PI3K inhibitor wortmannin32 did not affect LNO2-mediated protection, indicating no role for classical NO•/cGMP/PKG signalling, or ERK/PI3K signalling. Furthermore, post-SIR mitochondrial function (intracellular Δψm) correlated well with cell viability and benefited from LNO2 treatment (Figure 2B).
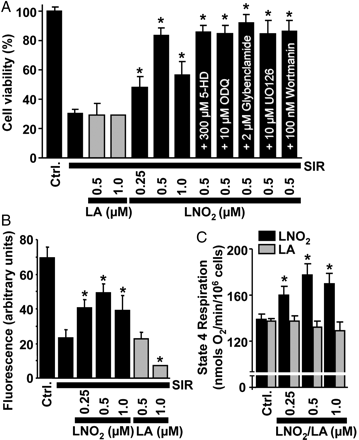
LNO2 protects cardiomyocytes from SIR injury and stimulates myocyte respiration. (A) Post-SIR cell viability. Cardiomyocytes were subjected to SIR injury in the presence of indicated concentrations of LNO2 or LA, added 20 min before ischaemia. Where indicated, the mKATP antagonists 5-HD or glybenclamide, the sGC inhibitor ODQ, the ERK inhibitor UO126, or the PI3K inhibitor wortmannin was present from the beginning of incubations. (B) Effects of LNO2 on post-SIR intracellular mitochondrial membrane potential, measured using TMRE fluoresence. Treatment groups were as in Figure 2A. (C) Effects of LNO2 or LA on cellular state 4 respiration rate (clamped with oligomycin). All data are means ± SEM, n > 5. *P < 0.05 vs. SIR alone in (A) and (B), or *P < 0.05 vs. control in (C).
We next investigated the mechanism of LNO2-mediated protection and hypothesized that LNO2, like other electrophilic lipids,23 may uncouple mitochondria, which itself is known to be cardioprotective.10,11,24 Consistent with this hypothesis, Figure 2C shows that LNO2 stimulated cellular state 4 respiration (a surrogate marker for uncoupling), while LA was without effect. Such respiratory stimulation could be due to uncoupling, or an acceleration of oxidative-phosphorylation,10 therefore, we next assayed the direct effects of LNO2 on uncoupling in isolated mitochondria. Titration curves of state 4 respiration vs. Δψm (Figure 3) showed that 1 µM LNO2 stimulated uncoupling, as indicated by a left shift of the curve (a more H+ permeable membrane necessitates faster respiratory chain activity to maintain a given Δψm10), while LA was without effect. Figure 3B and C shows that LNO2-induced uncoupling was inhibited by the ANT inhibitor CATr and the UCP inhibitor GDP. Neither inhibitor affected baseline function in the absence of LNO2 (Figure 3D).
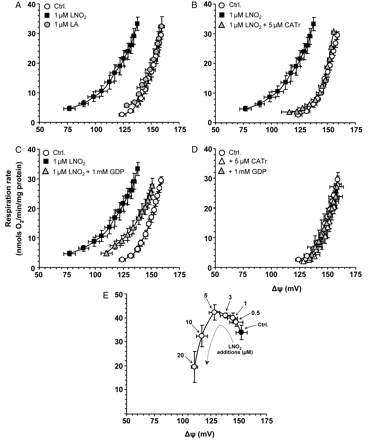
LNO2 stimulates mitochondrial uncoupling, in a manner sensitive to inhibitors of ANT and UCPs. Isolated mitochondrial uncoupling (H+ leak) was determined as previously.10 The upper-right point in each curve represents state 4 respiration, with the remaining curve resulting from titration with the complex II inhibitor malonate. An up/left shift in the curve indicates more uncoupled mitochondria. (A) Effect of 1 µM LA or LNO2. In (B) and (C), respectively, 5 µM CATr (ANT inhibitor) or 1 mM GDP (UCP inhibitor) was present where indicated, for 1 min before LNO2 addition. (D) Lack of effect of GDP or CATr on baseline (non-LNO2 stimulated) function. (E) Dose response to LNO2 of mitochondrial respiration and Δψm. The solid point represents state 4, with subsequent LNO2 additions (0.5, 1, 3, 5 µM) resulting in an up/left shift indicating uncoupling. Later LNO2 additions (10, 20 µM) resulted in a down/left shift indicating respiratory inhibition. All data are means ± SEM, n > 6.
Toxicity studies (Figure 3E) revealed that LNO2 >10 µM both inhibited respiration and dropped ΔΨm. This respiratory inhibition was not due to complex I inhibition (result not shown), as has been shown for other electrophilic lipids.33 Furthermore, similar to other electrophilic lipids,34 LNO2 >10 µM induced large-scale mitochondrial swelling indicative of PT pore opening (Figure 4). This effect was insensitive to the PT pore inhibitor cyclosporin A (CsA), indicating a possible role for the ‘unregulated’ PT pore resulting from membrane protein aggregation.35 PT pore opening was not induced by 5 µM LNO2 (Figure 4), suggesting this was not the mechanism of uncoupling induced by 1 µM LNO2 (Figure 3).
![Induction of unregulated PT pore opening at high [LNO2] (A) Typical PT pore swelling traces28 are shown. LNO2 or LA was added at the arrow. Where indicated, the PT pore inhibitor cyclosporin A (CsA, 2 µM) was present from the beginning of the experiment. (B) Quantitation of swelling magnitude at 10 min, from traces of the type shown in (A). Dose response to LNO2 is shown, with CsA effect shown for the 20 µM LNO2 dose (effect of CsA was similar at all LNO2 doses). Positive control for CsA-sensitive PT pore opening (to prove CsA was working appropriately) is shown on the right (open bars). Ca2+ was added at the arrow as shown in (A), instead of LNO2. All data are means ± SEM, n > 5.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/82/2/10.1093_cvr_cvn323/1/m_cvn32304.gif?Expires=1716515513&Signature=K1fw44cflRSbZTHTdb~gPkXrVIq7hBCHyd9Luiy~8zK0NrbFvy-oS4dkNwrkfhXHGRZI51VBatJXuNzWJ-HgnIPyXjEZd0zUerL5I4pfxyj1Vc7n6Dfeu9v3b~sSe1iRDdT3Oy6MDHqnRwjfZ1qKLTIfiQcR6S50cgLNbQETGAmFixwxZUGWGTxOV1j~QMs4cF8sE3H7O~zqaolIGvL5x7kbtqWB7FU3kjcAfbFlq69se4~2RV~DU-eRton68v2OU-VfGvGMndGQ0FzZNrVdOEDF72~kWhKG~oEL70Vh5UsAoCkFs1fZw87Jnuk04VVNkGacTXFEYzJ4nDG4q5Ml0A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Induction of unregulated PT pore opening at high [LNO2] (A) Typical PT pore swelling traces28 are shown. LNO2 or LA was added at the arrow. Where indicated, the PT pore inhibitor cyclosporin A (CsA, 2 µM) was present from the beginning of the experiment. (B) Quantitation of swelling magnitude at 10 min, from traces of the type shown in (A). Dose response to LNO2 is shown, with CsA effect shown for the 20 µM LNO2 dose (effect of CsA was similar at all LNO2 doses). Positive control for CsA-sensitive PT pore opening (to prove CsA was working appropriately) is shown on the right (open bars). Ca2+ was added at the arrow as shown in (A), instead of LNO2. All data are means ± SEM, n > 5.
Experiments to elucidate the mechanism of LNO2-induced uncoupling (Figure 5) employed oligomycin-clamped state 4 mitochondrial respiration as a surrogate marker for uncoupling.10 LNO2-induced uncoupling was not inhibited by BHT or c-PTIO, respectively, indicating no role for secondary lipid oxidation or NO• released from LNO236. However, LNO2-induced uncoupling was sensitive to the UCP-2 inhibitor genipin27 and was also reversed by E-SH suggesting protein thiol modification as a possible mechanism.16 Neither BHT, GDP, genipin, nor E-SH affected baseline state 4 respiration.
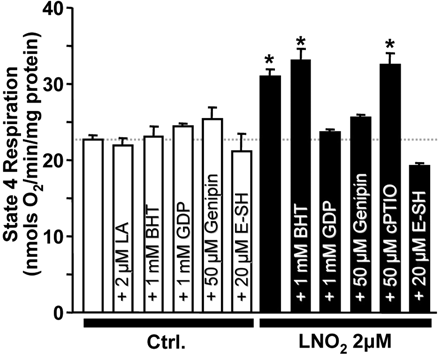
The effect of various reagents on LNO2-induced uncoupling. Oligomycin-clamped state 4 mitochondrial respiration was used as a surrogate for uncoupling (see Methods). Indicated concentrations of reagents were added prior to LNO2, except E-SH, which was added after LNO2 to reverse the latter's effects. Open bars represent effects of reagents in the absence or presence of native (non-nitrated) LA. Filled bars are in the presence of indicated concentrations of LNO2. GDP is a UCP inhibitor, genipin a UCP-2 inhibitor, E-SH a thiol reducing agent, c-PTIO a NO• scavenger, and BHT a lipophilic antioxidant. Data are means ± SEM, n > 6. *P < 0.05 vs. control.
To define mitochondrial targets of LNO2, biotin-tagged LNO226 was employed. Importantly, Bt-LNO2 induced mitochondrial uncoupling in the same GDP- and CATr-sensitive manner as native LNO2 (Figure 6A, B). Following Bt-LNO2 addition to mitochondria, biotinylated proteins were immunoprecipitated and western blotted with anti-biotin or anti-ANT antibodies. Figure 6C (upper panel) shows that Bt-LNO2 adducted several mitochondrial proteins, including a prominent band at ∼32 kDa which was identified as ANT (6C, lower panel). Furthermore, Bt-LNO2 labelled several proteins including ANT in intact cardiomyocytes (Figure 6D), thus indicating that Bt-LNO2, similar to other electrophilic lipids, can enter cells and target mitochondria.37 Full characterization of the nitroalkene-reactive proteome is anticipated to be the subject of subsequent studies. Nevertheless, as detailed in the Supplementary material online, several other mitochondrial and non-mitochondrial proteins were also identified as nitroalkylation targets.
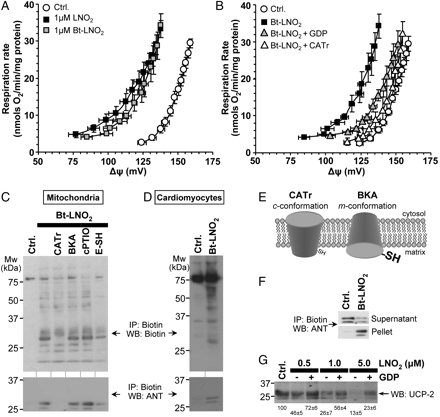
LNO2 nitroalkylates ANT and UCP-2. (A) Bt-LNO2 induced uncoupling similar to LNO2 (c.f. Figure 3), and (B) this effect of Bt-LNO2 was sensitive to GDP and CATr. Data are means ± SEM, n = 4. (C) Biotin labelled proteins were immunoprecipitated from Bt-LNO2 treated mitochondria and then western blotted with anti-biotin (upper panel) or anti-ANT (lower panel) antibodies. Ctrl. indicates mitochondria without Bt-LNO2. In all other lanes Bt-LNO2 (1 µM) was added. Where indicated 5 µM CATr, 20 µM BKA, 50 µM c-PTIO, or 20 µM E-SH was present. Inhibitors were added before LNO2, except E-SH, which was added after LNO2. Blots are representative of at least four experiments. (D) Similar experiment to (C), but with intact cardiomyocytes treated with Bt-LNO2, instead of mitochondria. (E) Schematic indicating the effects of ANT inhibitors CATr and BKA on ANT conformation. c cytosolic facing, m matrix facing. (F) Representative ANT western blot on a supernatant plus pellet pair from biotin immunoprecipitation of the type shown in (C). Upper panel is the IP supernatant (i.e. non-modified proteins which escaped pull-down) and lower panel is the pellet (i.e. Bt-LNO2 modified proteins pulled down by neutravidin). Data representative of four independent experiments. (G) Mitochondria were treated with indicated concentrations of non-biotinylated LNO2 in the absence or presence of the UCP inhibitor GDP (1 mM), and western blotted for UCP-2 (no immunoprecipitation). Blot is shown for an anti-N-terminal UCP antibody (Santa-Cruz), with similar results obtained using an anti-C-terminal UCP antibody (ADI, not shown). Representative blot from at least four independent experiments. Densitometry values (means ± SEM, as % of control) for the prominent UCP-2 band at 32 kDa are shown below the blot.
As shown in Figure 6E, two conformations of the ANT can be enforced by different inhibitors. Furthermore, in the CATr-induced c-conformation, a redox-sensitive thiol (C57) is inaccessible, whereas this thiol is exposed in the bongkrekic acid (BKA)-induced m-conformation.38 Support for this thiol as a potential target of LNO2 is provided by observations that CATr and E-SH inhibited Bt-LNO2 ANT modification, whereas BKA did not (Figure 6C). These results are consistent with the ability of both CATr and E-SH to inhibit LNO2-induced uncoupling (Figures 3B and 5). c-PTIO was without effect on ANT modification, indicating no role for NO• release. To quantify ANT modification, the fraction of ANT immunoprecipitated (i.e. ANT in pellet vs. supernatant) was examined. Figure 6F shows a pellet/supernatant pair from a biotin immunoprecipitation, western blotted for ANT. Densitometry on several such blots revealed that upon Bt-LNO2 treatment (1 µM) 48.2 ± 4.1% of ANT disappeared from the supernatant and appeared in the pellet.
Since LNO2-induced H+ leak was also sensitive to UCP-2 inhibitors (Figures 3C and 5), nitroalkylation of UCP-2 was also investigated. However, biotin-immunoprecipitation studies similar to those performed for ANT revealed no UCP-2 pull-down (data not shown). Subsequently, mitochondria were treated with native LNO2 in the presence or absence of the UCP inhibitor GDP, followed by western blotting for UCP-2. Figure 6G shows that UCP-2 progressively disappeared from the blot with increasing LNO2 doses, and this disappearance was attenuated by GDP. We hypothesized this may be due to nitroalkylation increasing the hydrophobicity of UCP-2 and preventing its SDS-PAGE migration. Supplementary material online, Figure S1D provides support for this hypothesis; blotting the entire gel including stacker plus comb revealed that high-LNO2 treatment resulted in aggregation of UCP-2 immunoreactivity at the stacker/separating gel interface.
4. Discussion
The major findings of this study are: (i) Electrophilic nitroalkene derivatives are formed endogenously in mitochondria during IPC; (ii) Exogenous LNO2 protects cardiomyocytes from SIR injury; (iii) LNO2 stimulates mitochondrial uncoupling, via ANT and UCP-2 dependent mechanisms; (iv) LNO2 nitroalkylates ANT and possibly UCP-2.
Central roles in IPC signalling are played by mitochondria,2–4 NO•,4–6 and ROS,18,19,24. However, the relationships between these key players are not fully understood. One unexplored mechanism for the interaction of ROS, NO•, and mitochondria in IPC may be the generation of electrophilic lipids such as nitroalkenes. Oxidative lipid derivatives are known to be cardioprotective39–41 and can induce mitochondrial uncoupling.23 Furthermore, mild mitochondrial uncoupling itself is cardioprotective.10,11,24 Therefore, the current data together with these previous findings suggest that electrophilic nitroalkenes may be endogenously generated in mitochondria during IPC and may induce mitochondrial uncoupling, thereby contributing to cardioprotection.
Regarding the contribution of this pathway to the overall cardioprotective effects of IPC, if a mitochondrial volume of 0.65 µl/mg protein is assumed,10 the amount of LNO2 in IPC mitochondria (Figure 1C) translates to an intra-mitochondrial concentration of 0.95 µM, which is the same level of exogenous LNO2 (1 µM) that stimulated uncoupling in isolated mitochondria (Figure 3). Thus, the mitochondrial concentration of LNO2 generated in IPC is theoretically capable of inducing uncoupling.
Considering the mechanism of acyl chain nitration in IPC, the finding that l-NAME only partially inhibited IPC-induced LNO2 formation (assuming efficient NOS inhibition by l-NAME in this system) suggests that non-NOS sources of RNS may be involved. In this regard, an observed ∼40% increase in mitochondrial NO2− levels during IPC (Nadtochiy and Brookes, unpublished) suggests NO2− may contribute to lipid nitration under the acidic conditions of IPC. The overall role of NOS in IPC is somewhat controversial,42 since in vivo studies suggest NOS is essential for IPC,43 whereas in vitro studies have found that l-NAME does not block IPC.44,45 The mechanism by which nitro fatty acids are liberated from membranes may involve PLA2, which is known to liberate linoleate and arachidonate, but not oleate, during IPC.46 This is consistent with our finding that mitochondrial OA-NO2 levels did not change in IPC, indicating some specificity in fatty-acid liberation.
Regardless the mechanism of nitroalkene formation or liberation in IPC, exogenous LNO2 was protective in a cardiomyocyte model of SIR injury. The signalling pathways by which nitroalkenes elicit protection could include PPARγ activation47–49 and subsequent HO-1 up-regulation.50 However, such gene transcription effects unlikely account for the immediate short-term (20 min) effects of LNO2 observed herein. Thus, we chose to focus on the short-term direct effects of LNO2 on mitochondrial function as a potential mechanism of protection and demonstrated that LNO2 induces mild mitochondrial uncoupling.
Since RNSs are known to have a number of other effects on mitochondria including the modulation of many proteins implicated in IPC,4 control experiments were performed to exclude the involvement of NO• released from LNO2, sGC, ERK, PI3K, or mKATP channels in LNO2-mediated protection. Furthermore, even though the mKATP channel has been proposed to uncouple mitochondria,18,31,51 the magnitude of mKATP flux is insufficient to account for IPC-induced uncoupling,10 and the current lack of a molecular identity for mKATP precludes its identification as an LNO2 target.
Having established that LNO2 uncoupled mitochondria, we next investigated the mechanism of uncoupling. One possibility is that cycling of protonated/deprotonated nitro-fatty-acids across the membrane, as observed for nitro-aromatics (e.g. dinitrophenol),52 could uncouple mitochondria. However, such uncoupling should not occur with Bt-LNO2 in which the carboxylic acid group is blocked by biotin. The data in Figure 6A and B thus precludes this mechanism. Rather, based on evidence from biotin-tagged LNO2 (Figure 6), the likely mechanism of LNO2-induced uncoupling is the nitroalkylation of ANT and UCP-2. Although the exact mechanism of H+ transport by these proteins is unknown,52 we speculate that nitroalkylation of cysteine residues may result in structural/conformation changes that cause uncoupling.
Mild mitochondrial uncoupling is thought to be cardioprotective via inhibition of mitochondrial Ca2+ overload2–4 or ROS generation,53 although we consider it unlikely54 that a direct role for UCPs in mitochondrial Ca2+ transport55 exists. Regarding ROS, it has been shown that uncoupling-induced cardioprotection is blocked by antioxidants, suggesting a role for ROS downstream of uncoupling.24 These apparently inconsistent findings are reconciled by the paradigm that low levels of ROS generated during IPC (prior to index ischaemia) may activate signalling processes that inhibit large- scale ROS generation during subsequent IR injury.10,19
In summary, the current work has elucidated components of a potential signalling pathway, in which nitroalkenes are generated in mitochondria during IPC and activate mitochondrial uncoupling via nitroalkylation of proteins such as ANT. These studies advance our understanding of the biological roles of nitroalkenes and also suggest that nitroalkenes may be useful cardioprotective pharmacologic agents. It is also possible that some of the cardioprotective benefits of NO2−56 or mitochondrially targeted NO• donors29 may involve nitroalkene generation, and that some cardioprotective benefits of the ‘Mediterranean diet’ may be due to intra-gastric nitroalkene generation.57
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: BAF acknowledges financial interest in Complexa, Inc.
Funding
This work was supported by grants from the US National Institutes of Health (RO1 HL071158 to P.S.B. and HL58115 and HL64937 to B.A.F.) and the American Diabetes Association (ADA 7-06-JF-06 to P.R.S.B).
References
Author notes
Authors contributed equally

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}