





Abstract
Coxsackievirus B3 (CVB3)-induced chronic myocarditis in mice is accompanied by severe fibrosis and by sustained elevation of platelet-derived growth factor (PDGF)-A, -B, and -C levels in the cardiac tissue. To test if PDGF stimulation of resident fibroblasts causally contributes to fibrosis, we employed inhibition of PDGF receptor signalling with the orally available kinase inhibitor Imatinib.
Chronic myocarditis was induced by CVB3 infection of major histocompatibility complex (MHC) class II knockout (B6Aa0/Aa0) mice. The mice were treated with 100 mg/kg Imatinib or vehicle, respectively, twice daily for 34 days. Expression of PDGF-C and of inflammatory cytokines were analysed by semi-quantitative RT–PCR. PDGFα receptor phosphorylation was detected by immunoblotting of cardiac tissue extracts and in situ by immunohistochemistry. Fibrosis formation was analysed by Sirius-Red staining and hydroxyproline (HP) determination. Fibronectin, and tenascin expression was analysed by RT–PCR and immunohistochemistry. Matrix metalloproteinase (MMP) activity was assessed with collagen, synthetic peptides, and gelatine as substrates. Imatinib significantly inhibited the myocarditis-related PDGFα receptor activation in the heart tissue. The virus titres in the hearts, inflammatory infiltrations, and elevated PDGF levels were unaffected by the Imatinib treatment. A significant attenuation of fibrosis occurred in Imatinib-treated animals. The Sirius Red-stained fibrotic area was reduced from 5.30 ± 0.50 to 3.21 ± 0.35%, and the HP content was reduced from 362 ± 43 to 238 ± 32 µMol/10 mg dry weight vs. 190 ± 27 in uninfected controls. The expression of fibronectin, EIIIA+ fibronectin, and tenascin C were likewise reduced. The diminished matrix protein deposition was not caused by elevated MMP activity, since MMP activity was not changed or even reduced under Imatinib.
The data suggest a causal role for elevated PDGF expression and PDGF receptor activity in the pathogenesis of cardiac fibrosis.
1. Introduction
Fibrosis is a common clinical problem. It can be observed in different organs, and is often associated with the final stages of chronic inflammatory processes.1–6 In the heart, extracellular matrix (ECM) components, notably fibrillar collagens of type I, III, and V, are important for tissue architecture and cardiac function. Endomysial collagen fibres around individual myocytes, perimysial fibres surrounding groups of myocytes and joining them with each other, and epimysial connective tissue are components of the physiological cardiac ECM.7,8 Collagen is normally stable, and synthesis and degradation occur by cardiac fibroblasts. Under pathological conditions, distinct cells designated myofibroblasts participate in collagen turnover. Both enhanced deposition of interstitial collagen and degradation of endomysial and permysial collagen contribute to cardiac dysfunction in hypertensive hearts.9 Excessive fibrosis, as it occurs under conditions of chronic myocarditis, can be classified as either replacement fibrosis, when functional tissue is replaced by connective tissue, or as reactive fibrosis, which is part of an adaptive process.8 In addition to collagen, tenascin C and fibronectin splice variants are often part of fibrotic tissue.10,11 Various cytokines and growth factors have been discussed to be involved in the causation of fibrosis, including TGFß,12 IL-1,13 TNFα,14 and platelet-derived growth factors (PDGFs).15 For fibrogenesis in the heart, members of the PDGF family are apparently important mediators. Transgenic overexpression of PDGF-C and PDGF-D, two more recently discovered PDGF isoforms,16 leads to massive cardiac fibrosis.15,17 We have previously shown that a strong upregulation of different PDGF isoforms, notably of PDGF-C, occurs in the hearts of Coxsackievirus B3 (CVB3)-infected major histocompatibility complex (MHC) class II knockout mice.18 This mouse model is considered as a system with relevance for the clinical setting since CVB3 is believed to play a role as a causative agent for the pathogenesis of chronic myocarditis, cardiac fibrosis, and dilatative myopathy in humans.19,20 Growth factors of the PDGF family exert their cellular actions by binding to receptors of the transmembrane protein-tyrosine kinase class, PDGFRα and PDGFRβ. These receptors dimerize, and the cytoplasmic tyrosine kinase is activated. PDGF-C selectively activates PDGFα receptor homodimers.16 Signalling activity of all PDGF receptors requires the receptor tyrosine kinase activity. Therefore, inhibition of PDGFR kinase with small molecules presents one possibility to evaluate the causal involvement of PDGFR activation in fibrogenesis. In this study, we took advantage of the possibility to inhibit PDGFR activity in vivo with the orally available tyrosine kinase inhibitor Imatinib mesylate (formerly designated STI571). This compound has been originally developed to block the pathogenic tyrosine kinase activity of BCR-ABL in the treatment of chronic myeloid leukaemia.21 It is, however, also a potent inhibitor of all PDGFR isoforms,22 and has been used earlier to demonstrate involvement of activated PDGF receptors in different pathogenic processes. Here we show that Imatinib inhibits activation of cardiac PDGF receptors, and effectively attenuates fibrosis in the CVB3-infected mice.
2. Methods
2.1 CVB3 infection
MHC class II knockout mice23 were bred under specific-pathogen-free conditions. They were held in groups of five in individually ventilated cages (IVC) under negative air pressure (−5 to −7 Pa). The CVB3 Nancy strain (CVB3-Mü/J, kindly provided by Prof. Kandolf, Department of Molecular Pathology, University Hospital Tuebingen, Germany) was propagated four times in HeLa cells. Adult 8–12-week-old males were inoculated intraperitoneally (i.p.) with 104pfu of CVB3-Mü/J as described.6,18 The mice were sacrificed under ether narcosis at the indicated time-points post-infectionem (p.i.). Anti-CVB3 IgM antibodies in serum were measured by ELISA at day 7 p.i.;6 non-responding mice were eliminated at this time. Virus titre determinations on HeLa cells6 were performed by the tissue culture infectious dose 50 (TCID50) assay method according to Reed and Muench24 with an additional group of animals (n = 19 treated with Imatinib, n = 15 treated with vehicle/water) at day 21.
2.2 Drug treatment protocol
CVB3-infected and sham-infected mice were treated two times daily with 100 mg/kg Imatinib by oral gavage with 12 h interval, or with water, from day 1 p.i. to day 34 p.i. Imatinib was provided by Novartis Pharma AG (Basel, Switzerland). The total number of animals indicated in Supplementary material online, Table S1 were treated in three independent experiments with 10–15 animals per treatment group. Analysis of fibrosis formation was done for all animals. Samples for analysis of the other parameters were randomly taken from the different experiments (see Supplementary material online, Table S1). Animals were sacrificed 12–14 h after the last Imatinib gavage. Cardiac tissue concentrations of Imatinib under these conditions exceeded the IC50 for PDGFR inhibition by at least one order of magnitude (data not shown; Heart samples were kindly analysed by Dr Gilles-Jaques Riviere, Novartis Pharma SAS, Rueil-Malmaison Cedex, France).
2.3 RNA extraction and RT–PCR
Total RNA was isolated according to Chomczynski and Sacchi.25 Reverse transcription was performed with 10 µg total RNA with oligo (dT) and 500 U MMLV reverse transcriptase (Superscript II, Invitrogen) according to the manufacturer's protocol. Fifty microlitres PCR reactions contained 0.2 mM of each dNTP, 20 nM of each primer, 1.25 U HotStar Taq polymerase, 5 µL 10× concentrated PCR buffer, 10 µL solution Q, and 2.5 mM MgCl2. Amplification was performed for 30 cycles according to the manufacturer's protocol. Primer pairs, and the lengths of the amplified products were as follows: β-actin, 586 bp (primer sequences refer Montgomery and Dallmann26), VP1, 851 bp (VP1 sense primer 5'-GGC CCA GTG GAA GAC GCG-3′, and VP1 antisense primer 5′-AAA TGC GCC CGT ATT TGT CAT TG-3′); PDGF-C, 395 bp (primer sequences refer Grün et al.18); total fibronectin, 531 bp (primer sequences refer Eismann et al.11); EIIIA+ fibronectin, 444 bp (primer sequence refer van der Straaten et al.27); tenascin C, 447 bp (tenascin sense primer 5′-GCA AGC TGA TCC AAA CCA TCT TCA CAA C-3′, and tenascin antisense primer 5′-AGT CAC CTG CTG TTC CAC TGT ATC C-3′28). The expression of precursor mRNA for the natriuretic peptide ANP was analysed with the primers 5′-CAG CAT GGG CTC CTT CTC CA-3′ (sense) and 5′-TCC ACT CTG GGC TCC AAT CCT-3′ (antisense), using 25 cycles for amplification.29 The amplified product was analysed on agarose gels. The stained bands were quantified by densitometry using Herolab E.A.S.Y. store Rev. 3.23 equipment and software (Herolab GmbH, Laborgeräte Wiesloch), and are given in arbitrary units normalized to β-actin. For each group of treatment, the measured values were expressed as mean±standard error of mean (SEM).
2.4 Detection of activated PDGFα receptors by immunoblottting
Twenty-to-thirty milligram samples of frozen (−80°C) cardiac tissue were mashed with a bead mill (Type MM 300, Retsch GmbH, Haan, Germany, frequency 30/s, for 2 min) using 200 µl lysis buffer containing, 50 mM Hepes pH 7.5, 1% Triton X-100, 0.15 M NaCl, 1 mM EDTA, 25 mM NaF, and freshly added 2 µg/mL leupeptin, 1% aprotinin, 2 µg/mL Pepstatin A, 1 mM PMSF, 2 µg/mL Pefablock (Roche, Mannheim, Germany), and 1 mM sodium orthovanadate. The homogenates were centrifuged at 15 000g for 20 min. Hundred micrograms of extracted protein was subjected to pulldown with wheat germ agglutinin beads (Sigma, Deisenhofen, 30 µl 1:1 suspension in lysis buffer) and the enriched glycoproteins were subjected to SDS–PAGE and immunoblotting consecutively using anti-phosphotyrosine antibodies (mAb 4G10, Biomol, Hamburg, Germany), and anti-PDGFRα antibodies (anti-PDGFRα (sc-951, Santa Cruz Biotechnology) for detection.
2.5 Measurement of collagenase and gelatinase activity
Frozen cardiac tissue was extracted as described earlier, however, with lysis buffer containing 50 mM Tris, pH 7.5, 0.5% Triton X-100, and 1 mM CaCl2. Fifty microgram protein of these samples were analysed using the Collagenase Assay Kit of Chondrex, Inc. (Redmond, USA) according to the instructions of the manufacturer with FITC-labelled bovine collagen I as substrate, and using 4-aminophenylmercuric acetate as collagenase activator. Gelatinase(s) were detected by zymography in SDS–PAGE gels, as described by Schurigt et al.30 As an alternative assay to quantify the activity of matrix metalloproteinases (MMPs) in cell lysates, the synthetic peptides Mca–Pro–Leu–Gly–Leu–Dap(Dnp)–Ala–Arg–NH2 (peptide 1) and Mca–Arg–Pro–Lys1Pro–Val1Gly–Nva–Trp–Arg–Lys(Dnp)–NH2 (peptide 2) (Bachem, Heidelberg, Germany) were used. Cell lysates were diluted to a final protein concentration of 0.8–1.2 mg/mL, then incubated with 5 µM substrate at 37°C in incubation buffer (100 mM Tris–HCl, 100 mM NaCl, 30 mM CaCl2, 0.05% Brij, 0.01% sodium azide, pH 7.6) for 2 h. Cleavage of the quenched substrates was detected as increase in fluorescence at 390 nm (λex = 330 nm).
2.6 Immunohistochemistry
Immunohistochemistry was performed as described before.18 Incubation with primary antibodies was carried out at 4°C overnight using rabbit anti-phospho-PDGFRα (Tyr745), 1:100; anti-PDGFRα (sc-951), 1:100; (all antibodies Santa Cruz Biotechnology), anti-phospho-Akt (Ser473) (Cell Signaling Technology, Inc., Cummings Centre Beverly, USA), 1:100; rabbit anti-chicken tenascin C (cross-reactive with murine tenascin C, Chemicon International Inc., Temecula, CA, USA), 1:200; biotinylated mouse anti-human EIIIA+ fibronectin (clone IST9, cross reactive with mice, biotinylation was done with the Animal Research Kit from Dako, Germany). Negative controls were done with 4 µg/mL rabbit IgG (SC-2027; Santa Cruz, Biotechnology) or with 4 µg/mL mouse IgG (MOPC-21; SIGMA). Sections were counterstained with Haemalaun.
2.7 Histological assessment of fibrosis
Samples of murine hearts were treated as reported previously.18 For quantification of Sirius-Red staining, we analysed the stained sections by a video system equipped with the image analysis system Quantimet 500/software QWin (Leica, Bensheim, Germany). The Sirius-Red stained area can thereby be quantified relative to the total tissue area. The analyses were performed with four consecutive sections taken from four different areas of each heart (16 analyses/heart).
2.8 Hydroxyproline detection
Heart samples were weighed, dried by lyophilization, and weighed again. The dried samples (9–25 mg dry weight) were transferred into a test tube, redissolved in 1 mL 6 M HCl, and hydrolyzed at 114°C for 24 h. The complete hydrolyzate was then transferred onto a column containing 200 mg Dowex W-X8(H)-resin, as well as 250 µl internal standard (3,4-dehydroproline ∼708 µmol/l), subsequently mixed with 1.5 mL phosphate/citrate buffer (pH 5.0, 0.2 M disodiumhydrogenphosphate, and 0.1 M citric acid; 10.3:9.7, V/V), and shaken at room temperature at 1000 r.p.m. for 10 min. After centrifugation at 5000 r.p.m. for 5 min, the supernatants were removed. The resin was resuspended in 6 mL H2O, vortexed vigorously, and centrifuged again at 5000 r.p.m. for 5 min. The supernatant was completely removed, and the closed tube with the resin was incubated at 115°C for 16 h. The samples were cooled to room temperature, mixed with 1 mL acetate/citrate buffer (pH 6.0, 58.7 g tri-sodium citrate-dihydrate, 57.1 g sodium acetate-trihydrate, and 5.5 g citric acid dissolved in 800 mL H2O), shortly vortexed, incubated at room temperature for 10 min, and finally centrifuged at 5000 r.p.m. for 5 min. Four hundred microlitres of the supernatant was diluted with 600 µL extraction buffer (80% methanol, 20% acetate buffer). HP was detected by an automatized HPLC method, relative to HP standards run in parallel (100, 300, and 500 µmol/L).31
2.9 Statistical analysis
All statistical analyses were performed using SPSS software (SPSS Inc., USA). Values are expressed as mean±standard error of mean (SEM). If not indicated differently, the Mann–Whitney rank sum test was used to compare each variable between different treatment conditions.
An overview of the number of mice entering the different types of analysis is given in Supplementary material online.
2.10 Ethical statement
The investigation conforms with the Guide for the Care und Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The experiments were approved by the local approval committee for animal experiments (permission number 02-01/04).
3. Results
3.1 Treatment with Imatinib inhibits PDGFR activity
CVB3 infected MHC class II knockout mice were employed to monitor the effect of Imatinib on the development of myocarditis and fibrosis. CVB3-infected animals were mock-treated or treated for 34 days with 100 mg/kg Imatinib twice daily. Control groups (sham-infected and mock-treated or Imatinib-treated) were run in parallel (Supplementary material online, Table S1).
We initially assessed the possibility that Imatinib would affect the virus replication and the CVB3-induced inflammatory process. Assays of CVB3 virus titres using the tissue culture infectious dose 50 (TCID50) method revealed that Imatinib treatment did not interfere with virus replication. Similar titres were detectable in the hearts of Imatinib-treated animals, and animals which received only vehicle (Supplementary material online, Figure S1A). Histology using HE stainings revealed a similar extent of inflammatory infiltrations under both conditions (Supplementary material online, Figure S1B and Table S2). In keeping with the ongoing inflammation under Imatinib-treatment, the levels of several inflammatory cytokines (including TNFα, IL-1β, IL-1α, and IL-6) analysed by semi-quantitative RT–PCR with RNA of the heart samples as described earlier18 were likewise not affected (data not shown). Since we had observed earlier,18 that PDGF-C levels are elevated in the CVB3-infected hearts, the expression of PDGF-C was assessed by semi-quantitative RT–PCR. Importantly, Imatinib treatment had no detectable effect on these elevated PDGF-C levels. Further, in agreement with unaffected virus titres, mRNA levels for the viral gene VP1 were also not altered under Imatinib treatment (Supplementary material online, Figure S1C). Taken together, the CVB3-mediated inflammation was not affected by Imatinib.
Elevated PDGF-C levels are expected to activate resident PDGFα receptors, and Imatinib treatment should interfere with this process. With an immunoblotting assay, phosphorylation of PDGF receptors in the CVB3-infected hearts and a significant inhibition by Imatinib treatment could be detected (Figure 1A). We also employed immunohistochemistry with antibodies recognizing the activated cognate receptor for PDGF-C, PDGFRα, and the activated downstream mediator of PDGFR signals, Akt, to qualitatively evaluate the status of PDGFR signalling. Consistent with the biochemical data, PDGFRα phosphorylation was elevated in the hearts of infected animals (Figure 1C). As reported earlier,18 PDGFRα levels were also increased in the infected hearts, and Akt phosphorylation was likewise elevated (Figure 1C). Under the treatment with Imatinib, PDGFRα phosphorylation, and activation of Akt were strongly reduced. Obviously, the drug reached effective levels in the hearts of the treated animals, and potently blocked PDGFR signalling.
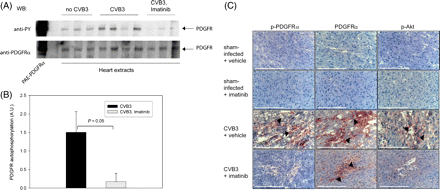
Imatinib inhibits PDGFR activity in the chronically inflamed heart tissue. Tissue extracts of cardiac tissue from CVB3-infected, or control sham-infected mice, treated with Imatinib or vehicle for 34 days as indicated, were analysed for phosphorylation of resident PDGF receptors by enrichment of PDGF receptors and immunoblotting with anti-phosphotyrosine antibodies (anti-PY) as described under Methods. An example blot is shown in (A), quantitative comparison of signals in tissues of CVB3-infected mock-treated (n = 7), or Imatinib-treated (n = 7) animals is shown in (B) (means±SEM). (C) Frozen myocardial sections of the indicated tissues were immunostained with antibodies to phosphorylated platelet-derived growth factor receptor α (p-PDGFRα), PDGFRα, or phospho-Akt (p-Akt), as indicated. The respective immunoreactive cells in inflammatory regions are indicated by black arrows. Sections were counter-stained with Haemalaun. Scaling bars correspond to 100 µm.
3.2 Imatinib inhibits the development of fibrosis
Cardiac fibrosis occurs in the CVB3-infected animals at day 21 p.i.,6 and is still progressive beyond this time-point.18 Histological analysis revealed that fibrosis occurring under these conditions was randomly distributed in the different heart regions, and was mainly replacement fibrosis. We hypothesized that inhibition of PDGFR signalling by Imatinib might attenuate fibrosis. We therefore chose to analyse fibrosis development at day 35 p.i. Histochemical analysis of fibrosis development was performed by Sirius-Red staining. A typical appearance of the connective tissue staining is illustrated in Figure 2A. Intense fibrosis development, as it occurs in the chronically infected hearts, was clearly attenuated in animals of the Imatinib-treated group. A quantitative analysis of the Sirius-Red staining of the sections by video imaging revealed a statistically significant (P < 0.05) reduction of fibrotic areas (Figure 2B). Similar findings were obtained by biochemically analysing the collagen deposition, determining hydroxyproline (HP). As shown in Figure 2C, a substantial amount of HP can be detected in the hearts of non-infected control animals. The HP content was, however, substantially increased in the hearts of CVB3-infected mice. Under treatment with Imatinib, a strong and statistically significant (P < 0.05) reduction of the HP tissue content was observed. The magnitude of reduction of this parameter even exceeded the Imatinib-mediated decrease of Sirius-Red staining (compare Figure 2B). We also determined the influence of Imatinib-treatment on the mRNA levels of tenascin C (Tn-C), total fibronectin (Fn), and the splice variant EIIIA+ fibronectin (EIIIA+ Fn)11,32,33 by semiquantitative RT–PCR. A clear reduction of mRNA levels for all three markers was observed in hearts of Imatinib-treated mice, compared with the levels in vehicle treated mice (Figure 3A). Imatinib-mediated downregulation of Tn-C and Fn levels could also be confirmed by immunohistochemistry (Figure 3B). These data indicate that inhibition of matrix protein production rather than accelerated degradation is the reason for the reduced amount of connective tissue under Imatinib. To further address this issue, MMP activities in the cardiac tissue were assessed using collagen, gelatine, or two synthetic peptides with different susceptibility to MMP isoforms as substrates. An elevated activity of MMPs has previously been shown to accompany CVB3-induced acute myocarditis, and to contribute to left ventricular dysfunction.34–36 In the chronically inflamed hearts, we could, however, only detect a trend of increased activity in collagenase activity and MMP activity towards peptide 1 compared with uninfected controls (Figure 4A and C). These activities were significantly reduced under Imatinib treatment. No differences in gelatinase activity, or MMP activity detected with peptide 2 were detectable (Figure 4B and D). Notably, Imatinib treatment did not increase proteolytic activity in any of the assays.
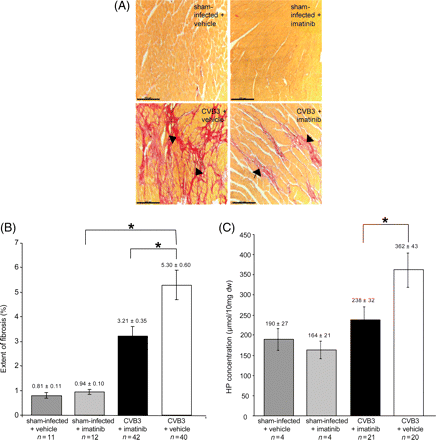
Imatinib inhibits fibrosis development in hearts of CVB3-infected mice. (A) Matrix protein deposition visualized by Sirius Red-staining. Paraffin-embedded tissue sections from hearts of CVB3-infected mice, and sham-infected mice, treated either with Imatinib, or with vehicle (as indicated) were stained with Sirius Red. Weak development of fibrosis (black arrows) was visible in Imatinib-treated CVB3-infected animals in comparison with extensive fibrotic areas in mice treated with vehicle only. Scaling bars correspond to 100 µm. (B) Quantitative analysis of matrix protein deposition detected by Sirius Red-staining. Sirius Red-stained areas were quantified by image analysis (given as % of total tissue area) in the indicated treatment group (means±SEM). Differences in the extent of fibrosis between CVB3-infected mice treated with Imatinib, and CVB3-infected mice treated with vehicle were statistically significant (Mann–Whitney test; P < 0.05*). (C) The HP content per dry weight was estimated in the cardiac tissue of the different treatment groups, as indicated (means±SEM). Statistically significant differences are indicated (Mann–Whitney test; P < 0.05*). The low number of samples for the two different groups of uninfected animals did not allow statistical comparison with the CVB3-infected animals. If, however, values for all sham-infected animals (vehicle- and Imatinib-treated) were combined, a statistically significant increase in HP content in CVB3-infected animals could be detected (362 ± 43 vs. 175 ± 17 in controls, P < 0.05).
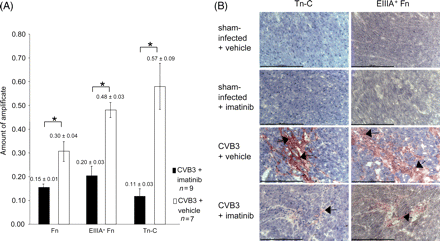
Imatinib inhibits the expression of matrix proteins in CVB3-infected hearts. (A) Effect of treatment with Imatinib on mRNA expression of total fibronectin, EIIIA+ fibronectin, and tenascin C in hearts of CVB3-infected mice. Semi-quantitative RT–PCR was evaluated by densitometric analysis (n = 4–9, mean±SEM). Statistically significant differences are indicated (Mann–Whitney test; P < 0.05*). (B) Frozen myocardial sections of CVB3-infected mice treated with Imatinib, or vehicle, respectively, from day 35 p.i. were reacted with antibodies to tenascin C (Tn-C), or EIIIA+ fibronectin (EIIIA+ Fn). Immunoreactive areas are indicated with black arrows. Sections were counterstained with Haemalaun. Scaling bars correspond to 100 µm.
![Imatinib treatment does not increase matrix metalloproteinase activity. Proteolytic activity was measured in cardiac tissue of uninfected, CVB3 infected, and CVB3 infected and Imatinib treated animals, as indicated. (A) Determination of collagenase activity with FITC-labelled collagen as substrate, (B) gelatinase activity assessed in zymograms, (C and D) proteolysis of fluorescently labelled peptides, which preferentially detect activities of MMPs 2, 3, 7, 8, 9, 10, 12, 13, 14, 17, 25, and 26 [peptide 1, (C)], or MMPs 3, 7, 9, and 12 [peptide 2 (D)] (n = 6, means±SEM).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/79/1/10.1093_cvr_cvn063/2/m_cvn06304.gif?Expires=1716480083&Signature=BGGBCGZoT07UibZL3s~iRex8Lh-CbY3KIuOTChn~8~DLjdfFjdOfyZy2LLHXBDg3R98mGNV~KtqMzsqoG2RothxJWQN6cKs-b28yFGhkfwBNYZq69IRAlzHeK70U43DK8Rq75yivtbRsnfjWzxrL97jwhYp1Oo4UvufkC9sW7Azf~ebyMwb9GKKTPxEd4FqLYOFR7MJXlsdrdxTROQme9m2SnlVcXKkLRmVpCU1k9wD0joUoORKS9o3IJ3Jovda1c3wopx9tOjXdIG3pFxmSQXCyhvyu8Q0hRhYPHHidmnW-R85uO1m-DmwmwuCj2mne0AuN20VgVnR4JsGWgjzE2Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Imatinib treatment does not increase matrix metalloproteinase activity. Proteolytic activity was measured in cardiac tissue of uninfected, CVB3 infected, and CVB3 infected and Imatinib treated animals, as indicated. (A) Determination of collagenase activity with FITC-labelled collagen as substrate, (B) gelatinase activity assessed in zymograms, (C and D) proteolysis of fluorescently labelled peptides, which preferentially detect activities of MMPs 2, 3, 7, 8, 9, 10, 12, 13, 14, 17, 25, and 26 [peptide 1, (C)], or MMPs 3, 7, 9, and 12 [peptide 2 (D)] (n = 6, means±SEM).
It has been shown that elevated expression of precursor mRNAs of natriuretic peptides notably ANP in cardiac tissue correlates with impaired cardiac function, for example in virally induced myocarditis,37 or myocardial infarction.38–40 To obtain initial data on possible effects of CVB3-induced chronic myocarditis and of Imatinib treatment on cardiac function, expression of the precursor mRNA of ANP was assessed by semi-quantitative RT–PCR. As shown in Figure 5, expression of ANP mRNA was increased in infected animals. Imatinib treatment had no significant effect on the increased expression level.
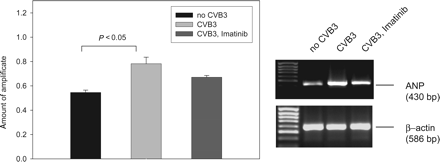
Expression of ANP precursor mRNA. Semi-quantitative RT–PCR was evaluated by densitometric analysis (n = 4–5, mean±SEM). A statistically significant difference in CVB3 infected animals from uninfected controls is indicated. The value in the presence of Imatinib was significantly different from the one in uninfected animals (P < 0.05), but not significantly different from the one in CVB3 infected, vehicle treated animals (P = 0.08). An example of the RT–PCR results is shown in the right panel (The lanes are from the same gel with identical processing, but were rearranged for better clarity.).
4. Discussion
Fibrosis is a hallmark feature of the chronic stage of viral myocarditis.5,19 The molecular mechanisms underlying fibrosis development in chronic myocarditis are currently not well understood. Several lines of evidence suggest that members of the PDGF family might be important for the development of cardiac fibrosis. Transgenic overexpression of PDGF-C and PDGF-D in murine heart led to massive cardiac fibrosis.15,17 In other fibrotic conditions, such as pulmonary, kidney, or liver fibrosis, factors of the PDGF family have been identified as important contributors to the fibrogenic process.1,41–44 We have previously found that chronic myocarditis in CVB3-infected MHC class II knockout mice is accompanied by sustained elevation of PDGF-A, -B, and -C levels in the cardiac tissue.18 Here we show that the PDGFR blocker Imatinib inhibits activation of resident PDGFR, and attenuates fibrosis in this mouse model significantly. These data strongly suggest that elevation of PDGF levels and subsequent activation of PDGFR causally contribute to the type of cardiac fibrosis which occurs in this model.
Efficacy of Imatinib for attenuation of fibrosis has recently been reported also for other organs.45–47 It can therefore be assumed that Imatinib-sensitive tyrosine kinases play a more general role in fibrogenesis.
In addition to PDGF receptors,46,48 Abelson tyrosine kinase (c-Abl) as a novel mediator of transforming growth factor beta (TGFβ)-signalling49 has been proposed as a relevant target for Imatinib in inhibition of fibrogenesis.45,50 While the data presented here, and data of others15,17 strongly suggest causal involvement of PDGFR in fibrogenesis, they do not allow to exclude that c-Abl activity is also involved in fibrogenetic signalling. It could function downstream of TGFβ receptors, but also partially mediate signalling of activated PDGFR.51 It is known that TGFβ can drive cardiac fibrosis when overexpressed in the mouse heart,52 and the possible contributions of the different TGFβ-mediated signalling mechanisms to cardiac fibrosis in our model of CVB3-induced chronic myocarditis remain to be investigated.
The CVB3 infection leads to activation of PDGFα receptors and activation of Akt. We assume that blocking of these events leads to inhibition of the proliferation of resident fibroblasts and myofibroblasts, and potentially also to inhibition of matrix protein production by resident fibroblasts. The dependence of matrix protein expression from PDGFR-mediated pathways in fibroblasts has been described.53,54 The reduced deposition of matrix proteins could be scored by the analysis of several parameters, including the measurement of Sirius-Red-stainable areas and the HP content of the tissue. Moreover, the expression of Tn-C, Fn, and EIIIA+ Fn was also reduced under the influence of Imatinib. Inhibition of the expression of the fibronectin splice variant EIIIA+ and of Tn-C may be of particular interest, since they are typically secreted by activated fibroblasts/myofibroblasts which play a prominent role in the development of fibrosis.32 Elevated activity of urokinase-type plasminogen activator and MMPs have recently been observed in the hearts of C57BL/6 mice 7 days after CVB3 infection. Inhibition of both types of proteinases prevented cardiac injury and dysfunction.35 Similarly, the reduction of MMP 2 and 9 mRNA expression by exogenous IL-4 treatment has recently been demonstrated to beneficially affect left ventricular function in acutely CVB3-infected mice.36 These findings highlight the need of the integrity of the physiological ECM for cardiac function, and indicate the possibility that modulation of proteolytic activities could also play a role for the reduction of fibrotic deposits which we have observed upon Imatinib treatment. Using different assay methods, we could detect MMP activity in the hearts of infected mice also at the analysed late time-points after CVB3 infection. However, they were not significantly higher than in uninfected animals, which may indicate that initially elevated MMP activity is again downregulated at the late time-points after infection which were analysed in this study. Importantly, Imatinib treatment did not elevate but even reduced the activity of those MMPs which were detectable with collagen or one of the employed fluorigenic peptide substrates. Although we cannot entirely rule out local increases in proteinase activity under Imatinib, our data suggest that the anti-fibrotic effect of Imatinib is indeed based on inhibited synthesis rather than enhanced degradation of deposited matrix proteins.
In our mouse model, Imatinib had little effect on the inflammatory process, indicated by the continuous presence of inflammatory infiltrations and inflammatory cytokines in the hearts of Imatinib-treated animals. It has previously been shown that CVB3-induced myocarditis impairs cardiac function in C57BL/6 mice.34,35 While we could not directly analyse cardiac function in the course of the experiments described here, the expression of precursor mRNA for the natriuretic peptide ANP was assessed as a surrogate marker of impaired cardiac performance. A CVB3-mediated increase of ANP mRNA precursor was not significantly affected in the Imatinib-treated mice. This observation is consistent with the ongoing inflammation in the cardiac tissue, and with the absence of effects of Imatinib on this process. These data suggest that inhibition of fibrosis by Imatinib may not be sufficient to improve impaired cardiac function in these chronically CVB3-carrying mice. Troponin I levels were elevated in the sera of CVB3-infected animals indicating cardiac tissue damage which was likewise unaffected by Imatinib treatment (Supplementary material online, Table S4). Further investigations are required to clarify the effects of Imatinib on cardiac function in CVB3-induced myocardits.
In conclusion, this study provides evidence for the causal involvement of activated PDGF receptors in the development of cardiac fibrosis in CVB3-induced chronic myocarditis. Imatinib treatment does not affect chronic inflammation, but is effective in attenuating fibrosis.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: E.B. is an employee of Novartis AG, Basel, Switzerland.
Funding
This work was supported by a grant from Novartis AG, Switzerland.
Acknowledgements
The authors thank Birgit Meißner for excellent technical work. B6Aa0/Aa0 MHC class II knockout mice were kindly provided by Professor Horst Bluethmann (Roche Center for Medical Genomics, Basel, Switzerland).
References
Author notes
Both authors contributed equally to this study.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}