





Abstract
Objective: This study investigated whether differences exist in atherogen-induced migratory behaviors and basal antioxidant enzyme capacity of vascular smooth muscle cells (VSMC) from human coronary (CA) and internal mammary (IMA) arteries.
Methods: Migration experiments were performed using the Dunn chemotaxis chamber. The prooxidant [NAD(P)H oxidase] and antioxidant [NOS, superoxide dismutase, catalase and glutathione peroxidase] enzyme activities were determined by specific assays.
Results: Chemotaxis experiments revealed that while both sets of VSMC migrated towards platelet-derived growth factor-BB (1–50 ng/ml) and angiotensin II (1–50 nM), neither oxidized-LDL (ox-LDL, 25–100 μg/ml) nor native LDL (100 μg/ml) affected chemotaxis in IMA VSMC. However, high dose ox-LDL produced significant chemotaxis in CA VSMC that was inhibited by pravastatin (100 nM), mevastatin (10 nM), losartan (10 nM), enalapril (1 μM), and MnTBAP (a free radical scavenger, 50 μM). Microinjection experiments with isoprenoids i.e. geranylgeranylpyrophosphate (GGPP) and farnesylpyrophosphate (FPP) showed distinct involvement of small GTPases in atherogen-induced VSMC migration. Significant increases in antioxidant enzyme activities and nitrite production along with marked decreases in NAD(P)H oxidase activity and O2− levels were determined in IMA versus CA VSMC.
Conclusions: Enhanced intrinsic antioxidant capacity may confer on IMA VSMC resistance to migration against atherogenic agents. Drugs that regulate ox-LDL or angiotensin II levels also exert antimigratory effects.
This article is referred to in the Editorial by D.G.M. Molin and M.J. Post (pages 3–4) in this issue.
1. Introduction
Coronary atherosclerosis, associated with oxidative stress, is the leading cause of morbidity and mortality in developed countries. Atherogenesis, a progressive process ultimately leads to formation of focal atherosclerotic plaques in the intima, characterized by foam cell formation and accelerated vascular smooth muscle cell (VSMC) proliferation and migration [1]. Low-density-lipoprotein (LDL)-cholesterol is a strong predictor of coronary artery disease and the HMG-CoA reductase inhibitors (statins) reduce the risk of primary and secondary events [2,3].
Statins are associated with other biological processes in the vessel wall, for example, simvastatin has been implicated in inhibiting platelet-derived growth factor (PDGF)-induced DNA synthesis in human glomerular mesangial cells [4]. PDGF secreted by macrophages exacerbates atherogenesis by stimulating VSMC proliferation and plaque neovascularisation [1]. Another non-lipid-lowering effect of statins is their ability to prevent migration of VSMC to PDGF by blocking isoprenoid production. Isoprenoids, like cholesterol, are HMG-CoA reductase pathway products and are required for prenylation of small monomeric GTPases, such as Ras and Rho, involved in cell migration control [5]. Hence, the inhibition of PDGF-induced mitogenesis and migration by statins should limit its deleterious effects.
Angiotensin II (Ang II) which exerts numerous effects on the cardiovascular system including the induction of VSMC migration, has also been implicated in atherogenesis [6]. Recent studies have revealed that angiotensin converting enzyme inhibitors (ACEI) reduce the risk of vascular diseases such as myocardial infarction and that Ang II type 1 receptor (AT1R) blockers (ARB) exert anti-atherosclerotic effects [7,8].
Ang II displays its proatherogenic effects through AT1R stimulation and as a consequence NAD(P)H oxidase enzyme activation to generate excess levels of superoxide anion (O2−), a reactive oxygen species (ROS) [9]. The effects of ROS range from oxidizing LDL and eliciting vasoconstriction to scavenging nitric oxide (NO), the most potent endogenous vasodilator that is produced from l-arginine by NO synthase (NOS) [10,11]. Under physiological conditions, O2− is converted to H2O2 by superoxide dismutases (SOD) which is then further metabolized to H2O by catalase and glutathione peroxidase (GPx) [12]. However, in pathological conditions, O2− may lead to atherosclerotic plaque formation via diminished concentrations and/or aberrant regulation of antioxidant enzymes.
Compared to coronary arteries (CA), human internal mammary arteries (IMA) rarely develop macroscopic atherosclerotic plaques despite their exposure to similar levels of atherogenic agents [13]. Therefore, IMA are considered as the best conduits for coronary bypass grafting. Indeed, IMA grafts have better patency rates compared to other arterial and venous grafts [13]. Although IMA are possibly not exposed to the same haemodynamic conditions and turbulence because of their anatomical location, it is reasonable to hypothesize that the absence of atherosclerotic plaques cannot solely be attributed to their location.
Since VSMC migration is a key factor in atherogenesis, the current study investigated first whether differences exist in the migratory patterns of CA versus IMA VSMC in the presence of agents associated with atherogenesis, namely native LDL, oxidized-LDL (ox-LDL), Ang II or PDGF-BB. Secondly, it investigated how cell migration was altered by free radical scavengers, molecules that specifically block receptors (CD36, AT1R and PDGF-βR), and statins. Thirdly, it explored the involvement of small monomeric GTPases to identify potential differences in CA and IMA VSMC migratory responses. Finally, it examined the putative differences in basal activity of prooxidant (NAD(P)H oxidase) and antioxidant (NOS, SOD, catalase and GPx) enzymes as well as nitrite and O2− production in these cell types.
2. Materials and methods
2.1. Patient characteristics
IMA samples were collected from patients (n=12) undergoing coronary bypass grafting. Patients with hypertension (>140/90 mm Hg), diabetes mellitus, hypercholesterolemia (>5.2 mM) and smoking within the past 6 months were excluded. Study was approved by the Research Ethics Committee of the Queen's University Belfast and protocol conformed to ethical guidelines of Declaration of Helsinki.
2.2. Human IMA VSMC isolation and culture
Under sterile conditions, the distal straight (area before the bifurcation) segments of human IMA were cleaned of adhering tissues and cut open longitudinally prior to denudation of endothelium and dissection of small squares (∼ 5 mm) to be placed for 30 min in smooth muscle growth medium 2 (SMGM2, Clonetics, UK) supplemented with antibiotic/antimycotic agents. Ten explants were placed with vessel lumens down in cell culture dishes previously wetted with 1.5 ml fetal calf serum. Subsequently 3 ml of SMGM2 was added into dishes and they were placed in a 37 °C incubator with growing surface uppermost. The dishes were inverted 3 days later and the culture medium was changed every 2 days.
To obtain normal coronary arteries from patients undergoing cardiac surgery is not ethically possible, hence normal i.e. genetically unaltered human CA VSMC (n=4 donors) were purchased from BioWhittaker (UK) and cultured as above in SMGM2. However, attention was paid to the donors' characteristics (age, sex and the cardiovascular disease) in order to match those of the patients from whom IMA cells were isolated. In this study, only VSMC between passages 4 and 8 were used.
2.3. Characterization of VSMC from human IMA and CA by immunocytochemistry
IMA or CA cells grown on coverslips were rinsed with ice-cold PBS and fixed in 4% formaldehyde for 20 min at room temperature before permeabilization with 0.1% Triton X-100. To characterize the cells as SMC they were stained with a monoclonal antibody raised against SM α-actin for 1 h in the dark at 4 °C prior to mounting in Vectashield and examination by confocal microscopy (Micro Radiance, Bio-Rad, UK). Staining of cells for von Willebrand factor failed to produce any staining and hence ruled out endothelial cell contamination (data not shown). Furthermore, the CA cells were previously stained positive for smooth muscle myosin, calponin and caldesmin by the commercial company.
2.4. Preparation of lipoproteins
Native LDL was isolated from human blood plasma by ultracentrifugation (density 1.019 to 1.063 g/ml) as previously described [14]. Oxidation of LDL was performed by exposure to CuCl2 (5 μM free Cu2+ concentration) in phosphate-buffered saline (PBS) at 37 °C for at least 24 h prior to detection of LDL and ox-LDL bands by lipid gel electrophoresis. Oxidation was terminated by the addition of EDTA (0.5 mg/ml) and LDL preparations were dialyzed by EDTA buffer. Native LDL and ox-LDL were kept in 50 mM Tris–HCl, 0.15 M NaCl and 2 mM EDTA at pH 7.4 and used within 10 days of preparation.
2.5. Migration assays
Cell migration was assessed by direct observation and recording of cell behavior in concentrations of recombinant human PDGF-BB (1–50 ng/ml; Insight Biotechnology Ltd., UK), Ang II (1–50 nM; Calbiochem), LDL or ox-LDL (25–100 μg/ml) using the Dunn chemotaxis chamber (Weber Scientific International, Teddington, UK). This apparatus permits the direction of movement of individual cells to be measured in relation to the direction of the gradient, as well as the time course to be followed. The apparatus was set up, filmed by time-lapse computer photography and data analyzed as described previously [15,16]. The rationale behind all chemotaxis experiments was the same; the potential chemoattractant was placed in the outer well of the Dunn chamber in addition to the components that were available in the inner well to rule out the contribution of those other components. Between the 2 wells cells on an inverted coverslip are exposed to the chemoattractant gradient and can be directly filmed for up to 24 h when the gradient will start to break down. For example, for LDL experiments SMBM was placed in the inner well and SMBM with varying concentrations (25–100 μg/ml) of ox-LDL or native LDL (100 μg/ml) in the outer well.
All cells were serum starved for 18 h prior to migration assays to exclude effects attributable to factors present in serum other than the expected chemoattractant. Experiments were run for 18 h with a time-lapse interval of 10 min. Minimum of 30 cells from three independent experiments were analyzed for each study. Where circular histograms are not shown (Figs. 2–5) the proportion of cells migrating towards the growth factor are given as a percentage of total cells in that field.
2.6. Inhibitor experiments
In those experiments where chemical inhibitors were employed the inhibitors were dissolved in DMSO and added to the medium to be used in both the inner and outer wells of the Dunn chamber (there was no period of pre-incubation with the inhibitors). The maximum final DMSO concentration was 1 μl/ml medium; this concentration was chosen for the control (solvent only) experiments. Inhibitors and concentrations used: The ACEI enalapril (1 μM, Merck, Sharp and Dohme), the statin pravastatin (100 nM), the statin mevastatin (10 nM), the rho kinase inhibitor Y-27362 (100 nM; all from Calbiochem), the ARB losartan (10 nM: Axxora UK Ltd.), the farnesyl transferase inhibitor (FTI) H-d-Trp-d-Met-p-chloro-d-Phe-Gla-NH2 (2 nM; Bachem, UK), the extracellular antioxidant Tiron (10 μM, Sigma), and the intracellular antioxidant MnTBAP (10 or 50 μM, A.G. Scientific).
2.7. Surface receptor antibody experiments
Cells were seeded on coverslips as described above and exposed to antibodies to the named surface receptors (CD36 or PDGF-β; R and D System, Minneapolis, MN) 16 h after the onset of serum starvation. The cells were then set up in the Dunn chamber 2 h later also in the presence of the surface receptor antibody. Antibody specificity was assessed by Western blot experiments of whole cell lysates which produced only the expected bands (data not shown).
2.8. Microinjection assay
Approximately 2×104 VSMC from CA or IMA were seeded in 25 mm Petri dishes containing 18-mm square No. 3 glass coverslips (Chance-Propper, UK) etched with a glass pen on the reverse side to facilitate localization of microinjected cells as described [5]. The cells on the coverslips were serum starved for 14 h and then exposed to the statin for 2 h before microinjection. For analysis of migration, all cells within the marked area were microinjected (InjectMan: Eppendorf AG, Germany) with control antibody (0.1 mg/mL) and either geranylgeranylpyrophosphate (GGPP) or farnesylpyrophosphate (FPP) peptides (all from Sigma). The microinjected coverslips were then set up in the Dunn chamber 2 h later still in the presence of the statin. At the end of the 18 h migration experiment microinjection confirmation studies were undertaken with immunofluorescent staining of control antibody as previously described [5,15]. Vehicle only microinjection had no effect on migration or cell viability.
2.9. Measurement of NAD(P)H oxidase activity and detection of O2− levels
O2− levels were measured by cytochrome C reduction assays. Briefly, the cells were collected in Hanks' balanced salt solution at a density of 20×106 cells/ml and incubated with 50 μM cytochrome C for 60 min at 37°C. O2− generation was measured as the superoxide dismutase (10 μg/ml)-inhibitable reduction of cytochrome C and monitored as the change in absorbance at 550 nm using a Cobas-Mira S centrifugal analyzer. Absorbances were recorded for 12 min with 90 s intervals and production of O2− was calculated as pmol O2− per 106 cells after subtracting background values measured at 550 nm. NAD(P)H oxidase activity was measured in similar experiments where the specific inhibitors of indicated ROS-generating enzymes, i.e. l-NAME (0.1 mM, eNOS), rotenone (50 μM, mitochondrial NAD(P)H oxidase), allopurinol (100 μM, xanthine oxidase) or indomethacin (50 μM, cyclooxygenase), were added to aliquots during 60 min incubation period prior to determining O2− generation.
2.10. Nitrite detection
Nitrite levels were measured in cellular homogenates using the Nitrate/Nitrite Colorimetric Assay Kit according to the manufacturer's instructions (Alexis Corporation, UK).
2.11. NOS activity assay
NOS activity was determined, in cell homogenates, by monitoring the conversion of [3H]l-arginine into [3H]l-citrulline with the NOS detect assay kit (Alexis Biochemicals, UK). Results were expressed as pmol l-citrulline/mg protein/min.
2.12. SOD assay
SOD activity was measured using a protocol dependent upon the inhibition in reduction of cytochrome C by endogenous SOD in cell homogenates. The superoxide anion, required for reduction, was generated by a reaction of xanthine–xanthine oxidase (XO). One unit of XO activity was defined by the amount of homogenate required to inhibit, by 50%, the rate of cytochrome C reduction. For total SOD assay, 0.1 mM xanthine was dissolved in 50 mM NaCO3, 0.1 M EDTA buffer. A dilute stock solution was added to a 10 μM solution of cytochrome C, 50 μM xanthine and 0.1 mM EDTA to produce a change in absorbance of 0.025/min at 550 nm at pH 10.
2.13. Catalase assay
The activity of catalase was determined in cell homogenates using a method adapted for automation on the Cobas Fara centrifugal analyzer [17]. Briefly, the activity was determined by monitoring the decomposition of H2O2 at 240 nm in the presence of methanol which produces formaldehyde which in turn reacts with Purpald (4-amino-3-hydazino-5-mercapto-1,2,4-triazole) and potassium periodate to produce a chromophore. Quantification was performed in comparison to the results that were obtained with catalase and formaldehyde standards.
2.14. Glutathione Peroxidase (GPx) assay
The activity of GPx was determined in cell homogenates using a colorimetric method [17]. Briefly, a fresh solution containing 0.3 U/ml glutathione reductase, 1.25 mM reduced glutathione and 0.19 mM NADPH in 50 mM Tris/HCl (pH 7.4) was prepared. Homogenates of 100 μg total protein were added to this solution and incubated for 3 min prior to addition of 12 mM t-butylhydroperoxide to commence the reaction. Absorbances were read at 340 nm for 4 min. Activities were calculated as nmol glutathione/mg protein/min.
2.15. Statistical analysis
To test for migration (chemotaxis) the Rayleigh test for unimodal clustering of directions was applied to the data and p<0.01 was chosen as the criterion for rejecting the null hypothesis of random directionality. Where there was significant unimodal clustering the mean direction and its 95% confidence interval were calculated and Tukey's post hoc analysis was conducted. Results for enzyme activity assays are presented as mean±SEM.; statistical analyses were performed by two-way analysis of variance (ANOVA) followed by Bonferroni–Dunn's post hoc analysis. p<0.05 was considered significant.
3. Results
3.1. Effects of LDL oxidation on migration of human CA and IMA VSMC
The response of CA and IMA to physiological and elevated levels of native and ox-LDL was investigated. Low concentrations (25 μg/ml) of ox-LDL produced random but not directed migration of CA or IMA cells (Fig. 1A and B respectively; p>0.05 for both). With increase in concentration (50, 75 or 100 μg/ml) ox-LDL had a differential effect on chemotaxis; that is, it produced directed migration in CA but not in IMA cells (cf. Fig. 1C, E and G with D, F and H). Native LDL (100 μg/ml) did not have a chemotactic effect on either type of VSMC (Fig. 1I and J). It would appear that it is not the level of LDL per se but oxidation which encourages VSMC migration in CA cells.
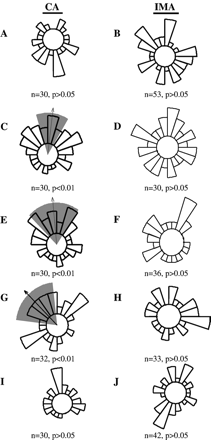
Migratory pattern of human CA and IMA VSMC in a gradient of ox-LDL or native LDL. Circular histograms show the proportion of cells migrating in each direction. The arrow indicates the mean direction of movement assessed by cyclical statistics (Rayleigh test: p<0.01). The grey wedge indicates the 99% confidence interval for the mean. Low levels of ox-LDL (25 μg/ml) did not induce chemotaxis in CA (A) or IMA (B) VSMC. The higher concentration of ox-LDL produced significant migration in CA VSMC (50, 75 and 100 μg/ml; C, E and G, respectively) but did not induce migration in IMA VSMC (D, F and H). Native LDL (100 μg/ml) did not induce migration in CA (I) or IMA (J) VSMC. n≥30 cells from 3 independent experiments were used.
3.2. Effects of ROS-scavengers on ox-LDL-induced migration of human CA VSMC
O2−, the foundation molecule of all ROS, may be involved in ox-LDL-mediated chemotaxis. Hence, the migratory properties of CA VSMC were investigated in the presence of either Tiron (10 μM: extracellular antioxidant) or MnTBAP (10 or 50 μM: intracellular antioxidant). Tiron or the lower dose of MnTBAP had no effect on migration to 50 μg/ml ox-LDL (Fig. 2). Only the higher dose of MnTBAP (50 μM) inhibited migration in ox-LDL-treated CA VSMC (p<0.01). Since ox-LDL is known to bind to the CD36 receptor, it was interesting to show that CD36 antibody could prevent chemotaxis to ox-LDL. The effect was receptor-specific since non-specific antibody from the same species and immunoglobulin subgroup as that for CD36 antibody could not mimic the response. O2− in VSMC is mainly generated by NAD(P)H oxidase in response to AT1R activation. Both losartan (10 nM), an ARB and enalapril (1 μM), an ACEI prevented migration to ox-LDL (Fig. 2). The migratory response of IMA cells to ox-LDL in the presence of antioxidants was not investigated since no migration occurred to ox-LDL in that cell type.
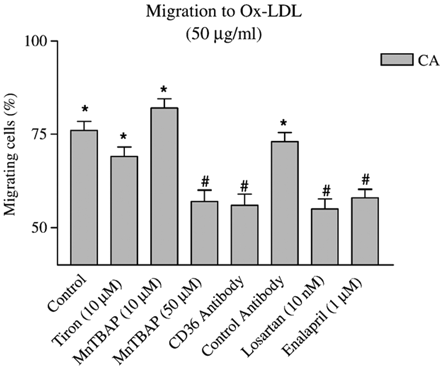
Effects of antioxidants (Tiron and MnTBAP), receptor blockade (Losartan, CD36 and its control antibody) and angiotensin-converting enzyme inhibitor (Enalapril) on oxidized-LDL-induced coronary artery smooth muscle cell migration. n≥30 cells from 3 independent experiments were used. *p<0.01 (Rayleigh test: chemotaxis detected); #p<0.01 (t-Test: % migrating cells vs. control).
3.3. Effects of Ang II on VSMC migration
Ang II has been shown to have pro-atherogenic effects, partly due to production of the free radical superoxide anion as a consequence of NAD(P)H oxidase activation [9]. The chemotactic dose response to Ang II (1–50 nM) was similar for CA and IMA (Fig. 3A). However, it is of interest that while extracellular antioxidant (Tiron) had no effect on chemotaxis, intracellular antioxidant (MnTBAP) had a differential effect on CA and IMA. A lower concentration of antioxidant (10 μM MnTBAP) inhibited Ang II-induced chemotaxis in IMA than in CA, where a higher concentration of MnTBAP was required to inhibit chemotaxis. Since Ang II signals mainly through the AT1R in VSMC, it was important to demonstrate that losartan prevented chemotaxis with Ang II (Fig. 3B).
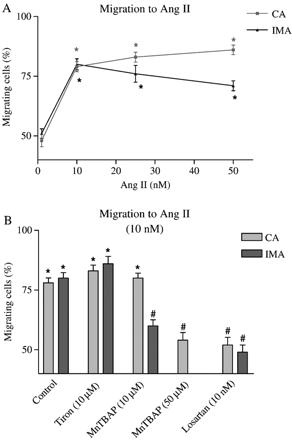
(A) Effect of Ang II on IMA and CA VSMC migration. (B) Effects of antioxidants (Tiron and MnTBAP) and Ang II type 1 receptor blocker (Losartan) on Ang II-mediated chemotaxis. n≥30 cells from 3 independent experiments were used. *p<0.01 (Rayleigh test); #p<0.01 (t-Test: % migration vs. cell type control).
3.4. Effects of PDGF-BB on VSMC migration
PDGF-BB is a well characterized chemotactic factor for VSMC. CA and IMA showed similar dose responses to PDGF-BB (1–50 ng/ml) with respect to chemotaxis (Fig. 4A). In contrast to ox-LDL and Ang II, antioxidants had no effect on PDGF-BB-induced VSMC migration in either CA or IMA. Since PDGF-BB signals mainly through the PDGF-β receptor in other VSMC [18], it was interesting to show that specific PDGF-β receptor antibody (but not non-specific control antibody) blocked PDGF-BB-induced chemotaxis in CA and IMA VSMC. The ARB losartan had no significant effect on PDGF-BB-induced chemotaxis (Fig. 4B).
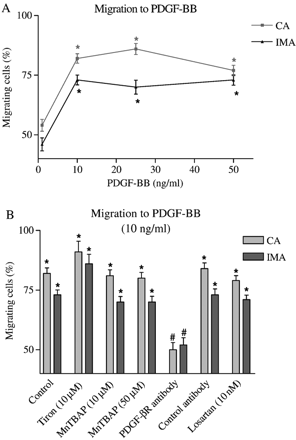
(A) Effect of PDGF-BB on IMA and CA VSMC migration. (B) Effects of antioxidants (Tiron and MnTBAP), Ang II type 1 receptor blocker (Losartan) and receptor blockade by specific and control antibodies on PDGF-BB-mediated chemotaxis. n≥30 cells from 3 independent experiments were used. *p<0.01 (Rayleigh test); #p<0.01 (t-Test: % migration vs. cell type control).
3.5. Effect of statins on CA and IMA VSMC chemotaxis
Mevastatin (10 nM: Fig. 5) or pravastatin (100 nM: data not shown) blocked chemotaxis to 50 μg/ml ox-LDL. To investigate if the inhibitory effect of mevastatin was due to alteration in the prenylation of small GTPases, isoprenoids were reintroduced to the cells by microinjection techniques. Both sets of cells were microinjected with either geranylgeranylpyrophosphate (GGPP; responsible for prenylation of Rho family small GTPases) or farnesylpyrophosphate (FPP; responsible for prenylation of Ras). In CA VSMC treated with mevastatin, chemotaxis to ox-LDL was restored by FPP but not GGPP microinjection. As IMA VSMC did not show directed migration to ox-LDL, the effects of GGPP and FPP were not assessed in these cells (Fig. 5A).
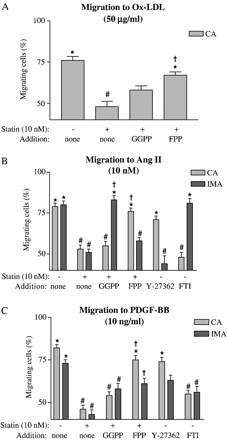
(A) Effect of oxidized-LDL on CA VSMC migration in the absence and presence of isoprenoids, namely geranylgeranylpyrophosphate (GGPP) or farnesylpyrophosphate (FPP). Effects of isoprenoids, Y-27362 (a Rho-kinase inhibitor) or FTI (a farnesyltransferase inhibitor) on IMA and CA VSMC migration in the presence of Ang II (B) or PDGF-BB (C). n≥30 cells from 3 independent experiments were used. *p<0.01 (Rayleigh test); #p<0.01 (t-Test: % migration vs. cell type control); †p<0.01 (t-Test: % of migrating cells vs. statin alone for that cell type).
Ang II-induced chemotaxis was blocked by mevastatin in both cell types. These effects were shown to be dependent on Ras and Rho prenylation in CA and IMA VSMC, respectively. To confirm these results migratory responses to Ang II were investigated using either a Rho-kinase inhibitor (Y-27362) or a farnesyltransferase inhibitor (FTI) in that while FTI prevented CA VSMC migration, Y-27362 blocked migration in IMA cells (Fig. 5B).
Mevastatin also inhibited migration of both cell types to PDGF-BB. CA VSMC, as was the case with ox-LDL and Ang II, had chemotaxis restored when microinjected with FPP indicating a role for Ras prenylation. A Ras-dependence in CA VSMC migration was again confirmed with the FTI being able to prevent migration to PDGF-BB. In contrast, IMA cells did not have chemotaxis restored in the presence of either GGPP or FPP (Fig. 5C). Since neither small GTPase alone restored chemotaxis, it was deemed possible that they might act synergistically. Indeed, microinjection of both GGPP and FPP into IMA cells in the presence of mevastatin restored directed migration to PDGF-BB (percentage migrating cells 80%, p<0.01).
3.6. Intrinsic antioxidant levels in human CA and IMA VSMC
Significantly higher levels of antioxidant enzyme activities along with a marked lower NAD(P)H oxidase activity were detected in IMA VSMC when compared with CA VSMC (p<0.05, Table 1). The differences in O2− and nitrite, the stable end-product of NO, levels were reflective of the differences observed in NAD(P)H oxidase and NOS enzyme activities, respectively (p<0.05).
Basal levels of nitrite and O2− and pro-oxidant and anti-oxidant enzyme activities in human CA and IMA VSMC
| CA VSMC | IMA VSMC | |
|---|---|---|
| Nitric oxide synthase (NOS, pmol l-citrulline/mg protein/min) | 0.39±0.09 | 1.56±0.25* |
| Glutathione peroxidase (GPx, U/mg protein) | 20.3±1.2 | 43.6±3.1* |
| Superoxide dismutase (SOD, mU/mg protein) | 99±5 | 142±26* |
| Catalase (U/mg protein) | 210±13 | 264±30* |
| NAD(P)H oxidase (pmol O2−/mg protein) | 1.33±0.16 | 0.42±0.11* |
| Nitrite (nmol/mg protein) | 19.99±1.23 | 34.42±1.28* |
| Superoxide anion (O2−, pmol/million cells) | 329±34 | 237±18* |
| CA VSMC | IMA VSMC | |
|---|---|---|
| Nitric oxide synthase (NOS, pmol l-citrulline/mg protein/min) | 0.39±0.09 | 1.56±0.25* |
| Glutathione peroxidase (GPx, U/mg protein) | 20.3±1.2 | 43.6±3.1* |
| Superoxide dismutase (SOD, mU/mg protein) | 99±5 | 142±26* |
| Catalase (U/mg protein) | 210±13 | 264±30* |
| NAD(P)H oxidase (pmol O2−/mg protein) | 1.33±0.16 | 0.42±0.11* |
| Nitrite (nmol/mg protein) | 19.99±1.23 | 34.42±1.28* |
| Superoxide anion (O2−, pmol/million cells) | 329±34 | 237±18* |
CA, coronary arteries; IMA, internal mammary arteries; VSMC, vascular smooth muscle cells. Data are expressed as mean±SEM from 4 different experiments. *p<0.05 difference between IMA and CA VSMC.
Basal levels of nitrite and O2− and pro-oxidant and anti-oxidant enzyme activities in human CA and IMA VSMC
| CA VSMC | IMA VSMC | |
|---|---|---|
| Nitric oxide synthase (NOS, pmol l-citrulline/mg protein/min) | 0.39±0.09 | 1.56±0.25* |
| Glutathione peroxidase (GPx, U/mg protein) | 20.3±1.2 | 43.6±3.1* |
| Superoxide dismutase (SOD, mU/mg protein) | 99±5 | 142±26* |
| Catalase (U/mg protein) | 210±13 | 264±30* |
| NAD(P)H oxidase (pmol O2−/mg protein) | 1.33±0.16 | 0.42±0.11* |
| Nitrite (nmol/mg protein) | 19.99±1.23 | 34.42±1.28* |
| Superoxide anion (O2−, pmol/million cells) | 329±34 | 237±18* |
| CA VSMC | IMA VSMC | |
|---|---|---|
| Nitric oxide synthase (NOS, pmol l-citrulline/mg protein/min) | 0.39±0.09 | 1.56±0.25* |
| Glutathione peroxidase (GPx, U/mg protein) | 20.3±1.2 | 43.6±3.1* |
| Superoxide dismutase (SOD, mU/mg protein) | 99±5 | 142±26* |
| Catalase (U/mg protein) | 210±13 | 264±30* |
| NAD(P)H oxidase (pmol O2−/mg protein) | 1.33±0.16 | 0.42±0.11* |
| Nitrite (nmol/mg protein) | 19.99±1.23 | 34.42±1.28* |
| Superoxide anion (O2−, pmol/million cells) | 329±34 | 237±18* |
CA, coronary arteries; IMA, internal mammary arteries; VSMC, vascular smooth muscle cells. Data are expressed as mean±SEM from 4 different experiments. *p<0.05 difference between IMA and CA VSMC.
4. Discussion
The major conclusions to be drawn from the current study are that the enhanced intrinsic capacity of antioxidant enzymes along with reduced bioavailability of O2−, through diminished activity of prooxidant NAD(P)H oxidase enzyme, helps to explain the relative resistance of human IMA VSMC to atherogenic agents-evoked migration compared to their counterparts from CA. Free radical scavengers that target intracellular O2− inhibit selectively Ang II- and ox-LDL-induced but not PDGF-BB-induced chemotaxis of CA and IMA VSMC. Moreover, statins, ACEI and ARB suppress VSMC migration in addition to their well-known effects related to cholesterol-lowering, Ang II synthesis and Ang II function. The statins attenuate chemotaxis by preventing prenylation of the small monomeric GTPases involved.
LDL-cholesterol accumulates in macrophages residing in the vascular wall and leads to the formation of foam cells and fatty streaks [19]. In vivo, oxidative modification of LDL occurs in endothelial cells, VSMC and macrophages within the arterial wall and subendothelial space [20]. Both native LDL and ox-LDL regulate the expression of chemotactic factors and cell adhesion molecules and reduce NO bioavailability, the most potent endogenous vasodilator, and thus lead to atherosclerosis in a time- and concentration-dependent manner [21]. Since VSMC migration in the arterial wall is a key feature in atherogenesis, the difference in migratory pattern of VSMC derived from CA and IMA to ox-LDL may in part account for the resistance of IMA to atherosclerotic disease.
The present study revealed that while native LDL (100 μg/ml) or low dose ox-LDL (25 μg/ml) had no impact on VSMC chemotaxis, higher concentrations of ox-LDL (50–100 μg/ml) produced a selective migration of CA VSMC. The migration of CA VSMC was abolished when the ox-LDL containing medium was supplemented with statins, losartan or enalapril. These findings support previous studies showing that (i) higher levels of plasma ox-LDL are required for the pathogenesis of CA disease and that oxidation of LDL enhances its atherogenic capacity [20]; (ii) statins inhibit coronary VSMC migration to lysophosphatidylcholine, a component of ox-LDL [22]; (iii) lowering serum LDL-cholesterol reduces the risk of cardiovascular events [2,3]; (iv) ARB suppress atherogenesis through reducing coronary VSMC migration [8]; and (v) ACEI reduce the risk of vascular diseases including myocardial ischemia [7].
Recent evidence implicate enhanced oxidative stress status (i.e. excess generation and/or release of ROS, in particular O2−) in the pathogenesis of coronary atherosclerosis; ROS may have direct vasoconstrictor effects, have the ability to scavenge NO reducing its bioavailability, and stimulate VSMC migration [10,11,23]. Incubation of VSMC with LDL is associated with excess production of O2− in a reaction that is coupled to induction of the AT1R subtype and subsequently activation of the NAD(P)H oxidase enzyme system [9,24]: that migration to ox-LDL could be blocked by antibody to its main receptor (CD36), an AT1R antagonist (losartan) and a O2− scavenger (MnTBAP) in the present work supports those findings.
Circulating levels of Ang II, the AT1R specific cytokine, are raised in atherogenesis and may potentiate CA and IMA VSMC motility [6]. PDGF-BB is a powerful mitogenic growth factor, which is also raised in the environs of a developing atheroma. Both Ang II and PDGF-BB produced significant chemotaxis in these cell types in the presence of physiological or elevated concentrations for each agent (≥10 nM and 10 ng/ml, respectively). The Ang II-evoked migration depended on AT1R activation, as losartan, an ARB, prevented migration in both IMA and CA VSMC. In contrast, migration to PDGF-BB depended on PDGF-β receptor binding and was losartan-insensitive for both cell types.
The reaction between O2− and NO generates another ROS termed peroxynitrite that oxidizes lipoproteins and contributes to foam cell formation in atherosclerotic lesions [25]. Under physiological conditions, SODs convert O2− to H2O2 that in turn is metabolized to H2O by catalase and GPx [12]. In pathological conditions this enzymatic pathway may be perturbed thereby leading to atherogenesis. The current study has for the first time revealed that IMA VSMC possess significantly greater levels of SOD, catalase, GPx and NOS enzymatic activities compared to their coronary counterparts. Indeed, the increase in NOS activity was mimicked by an increase in the levels of nitrite, the stable end-product of NO, in IMA versus CA cells. The elevated activity of NOS in IMA VSMC is critically important in that an adequate quantity of NO is required not only to maintain normal vascular tone but also to suppress VSMC proliferation and migration [26]. GPx, on the other hand, exerts beneficial functions by removing H2O2 and detoxifying the lipid hydroperoxides. A previous study investigating glutathione-related antioxidant defenses in carotid atherosclerotic plaques of 13 patients subjected to endarterectomy and in normal IMA of 13 patients undergoing CA bypass grafting showed that GPx was undetectable in the plaques of 7 patients and was considerably lower in the others compared to IMA [27]. Furthermore, increased GPx, Mn-SOD and catalase activities are associated with intracranial artery resistance to hypercholesterolemia-induced fatty streak formation compared to extracranial arteries, in human fetuses [28]. The significance of these data was emphasized by our findings revealing a diminished NAD(P)H oxidase activity and thus O2− generation in IMA versus CA VSMC. Considering the involvement of NAD(P)H-mediated overproduction of O2− in VSMC migration [23], it is plausible that reduced NAD(P)H oxidase activity and thus O2− production may contribute significantly to the resistance of IMA to atherosclerosis.
In the present study, the involvement of O2− production in Ang II-, PDGF-BB-or ox-LDL-induced cell migration was investigated through O2− scavengers, namely MnTBAP (10–50 μM) and Tiron (10 μM). It has been shown that while the lower concentration of MnTBAP, a cell-permeable SOD mimetic, is sufficient to inhibit Ang II-mediated IMA VSMC migration, higher quantities are required to inhibit those from CA in the presence of Ang II or ox-LDL. The studies show that the previously determined effective dose of Tiron, an exogenous O2− scavenger, failed to suppress migratory responses in either cell type in the presence of any of the atherogenic agents [29]. Taken together, these data imply that neutralization of endogenous O2− is a prerequisite for efficacious inhibition of VSMC migration and support our findings that IMA VSMC possess higher levels of SODs thereby requiring substantially less MnTBAP to eradicate atherogen-mediated migration. Reduced oxidative stress had no benefit for PDGF-BB-induced migration supporting our previous work in aortic VSMC migration studies [18]. These data confirm the results generated by losartan experiments that the elements which regulate PDGF-activated VSMC migration are different from those induced by ox-LDL and Ang II.
In the current study CA and IMA VSMC migration induced by ox-LDL, PDGF-BB or Ang II was inhibited by two statins, i.e. pravastatin and mevastatin. Statins lower cholesterol levels and inhibit isoprenoid intermediates (GGPP and FPP) synthesis that post-translationally modify (i.e. isoprenylate) proteins including the small GTPases Rho and Ras to facilitate lipid attachment with resultant functional activation [30]. To identify the mechanism(s) that account for migration-suppressive effects of statins, VSMC were microinjected with GGPP or FPP to recover directed migration in response to ox-LDL, Ang II or PDGF-BB. FPP and GGPP microinjection abolished inhibitory effects of statins selectively in CA and IMA VSMC, respectively. However, while FPP-mediated recovery of CA cell chemotaxis was independent of the atherogenic agent used, GGPP was only effective in IMA VSMC exposed to Ang II. These findings were corroborated by experiments employing a farnesyltransferase inhibitor (FTI) that blocks Ras/MAPK farnesylation and Y-27362, an inhibitor of Rho-kinase that prevents geranylated Rho activating its target Rho-kinase. While FTI significantly attenuated CA VSMC chemotaxis in the presence of Ang II or PDGF-BB, Y-27362 did not affect the migratory ability of these cells. In contrast, Y-27362 inhibited Ang II-induced IMA VSMC migration without affecting those stimulated by PDGF-BB. Moreover, in IMA VSMC both inhibitors suppressed chemotaxis to PDGF-BB. This suggests that the inhibition of HMG-CoA reductase through prevention of the isoprenylation of members of Ras and Rho families of low-molecular-weight GTP-binding proteins and is associated with the beneficial effects of statins. The critical roles of Rho, Ras and other small GTP-binding proteins including p21 Rac in the assembly of NAD(P)H oxidase enzyme system, cell proliferation, eNOS activity and thus NO generation, may shed additional light onto why IMA are relatively resistant to macroscopic atherosclerotic plaque development [31,32].
In conclusion, statins, free radical scavengers, ARB and ACEI may suppress the early events in atherogenesis by markedly inhibiting VSMC migration. The signaling pathways that are involved in statin-mediated suppression of migration appear to differ according to the vascular tree that the VSMC originate from. The enhanced antioxidant enzyme activities along with diminished NAD(P)H oxidase activity partially explain the resistance of human IMA VSMC to ox-LDL-induced chemotaxis. Given the observed resistance of IMA VSMC to ox-LDL and PDGF-BB-evoked migration, it is reasonable to suggest that the choice of a by-pass conduit should be with a tissue source able to resist atherogenesis in the persistent dyslipidaemic and growth factor rich environment.
Acknowledgements
This study was supported by a grant from Northern Ireland Chest Heart and Stroke Association. We thank Drs. J. McEneny and W.E. Allen, Queen's University Belfast and the surgeons in the Department of Cardiac Surgery, Royal Victoria Hospital for help and advice.
References
Author notes
Contributed equally to the manuscript.
Available online 27 June 2006 Time for primary review 32 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}