





Abstract
Objective: Reperfusion after ischemia may contribute to loss of myocardial function and increase in infarct size. Scavenging of reactive oxygen species (ROS) by glutathione (GSH) and inhibition of the sodium-proton-exchanger by cariporide are both capable of reducing myocardial reperfusion injury. We tested the efficacy of both agents applied regionally into the myocardium immediately before reperfusion. Methods: Neonatal rat cardiomyocytes (NRCMs) were exposed to either hypoxia (H, 8 h)/reoxygenation (R, 1 h) or H2O2 (300 μM) in the presence or absence of GSH (10 mg/ml). In pigs (n=5 per group), percutaneous LAD occlusion was performed for 60 min. Application of GSH (250 mg/kg) and/or cariporide (1 mg/kg) was achieved by pressure-regulated retroinfusion of the anterior cardiac vein draining the ischemic area starting 5 min before reopening of the occluded LAD. Seven days later, subendocardial segment shortening (SES) was analyzed by sonomicrometry. Infarct size was determined by methylene-blue staining of the non-ischemic area and tetrazolium red staining of the viable myocardium in the area at risk (AAR). Results: NRCM incubated with GSH (10 mg/ml) survived H/R or H2O2 (0.3 mM) to a larger extent than untreated cells. In pigs, infarct size of untreated hearts (51±6% of the AAR) was not significantly altered by GSH or cariporide retroinfusion alone (41±3% and 42±6%). In contrast, combined retroinfusion of cariporide and GSH significantly reduced infarct size (29±3%). SES of the infarcted area was improved only after cariporide/GSH retroinfusion as compared to untreated hearts. Additional systemic application of CD18-antibody IB4 (1.5 mg/kg) did not alter infarct size or SES in comparison to GSH/cariporide retroinfusion alone. Conclusion: Timely application of GSH scavenging ROS and cariporide targeting ion imbalance provides cardioprotection to the postischemic heart, which is superior to either treatment alone. The lack of an effect of additional IB4 treatment may indicate that GSH/cariporide retroinfusion itself affects leukocyte-dependent reperfusion injury.
1 Introduction
Reperfusion of an occluded coronary artery, established as standard treatment in the case of myocardial infarction, may induce arrhythmias [1], myocardial stunning [2] and microcirculatory flow disturbances such as no reflow [1,3] in the postischemic myocardium. Cellular mechanisms mediating these pathophysiological phenomena include formation of reactive oxygen species (ROS) [4] by myocytes [5], endothelial cells [6] and adherent leukocytes [7]. In addition, myocyte ion imbalance leading to e.g. calcium overload and hypercontracture [8], is involved in the loss of postischemic myocardial organ function. Consistently, therapeutic approaches to prevent or reduce myocardial reperfusion injury included application of reactive oxygen species scavengers and sodium-proton-exchanger inhibitors (NHE-I).
However, enzymatic elimination of reactive oxygen species by superoxide dismutase and catalase lacked in vivo efficacy in preclinical large animal models [9–11] although myocyte protection was achieved in vitro [12]. Alternatively, supplementation of physiological ROS scavengers such as GSH is an attractive option, since intracellular GSH levels decrease rapidly after ROS-exposure. In isolated perfused rat hearts, GSH supply during ischemia offers cardioprotection during reperfusion, as long as a certain length of ischemia is not exceeded [13]. For this purpose, however, high concentrations of GSH are required, e.g. 5 mM in an isolated rat heart model [13] or 800 mg/kg in a pig model [14]. GSH supplementation at this scale appears difficult to achieve in patients, a limitation potentially overcome by targeted application, such as pressure-regulated retroinfusion into the ischemic myocardial region.
Beyond ROS-induced detriment, evidence suggests that myocyte ion imbalance is a major cause of myocardial ischemia–reperfusion injury. As demonstrated in adult cardiomyocytes [15], the sodium hydrogen exchanger (NHE) inhibitor cariporide protects against post-hypoxic myocyte contracture. In addition, several animal studies revealed a cardioprotective effect of NHE inhibitors in large animal models when applied before or early in the course of ischemia [16–18], as opposed to application at the onset of reperfusion [18]. In contrast, systemic application of NHE-inhibitors in patients with acute coronary syndromes did not improve postischemic myocardial infarct size or patient outcome [19,20].
Taken together, despite experimental data suggesting an effect of either scavenging reactive oxygen species or attenuating ion imbalance in the course of myocardial ischemia–reperfusion, the limited efficacy of systemic application seems to hinder a detectable success in clinical trials. Recently, we demonstrated that selective, pressure-regulated retroinfusion is a efficient regional approach of drug, gene or protein application into the ischemic myocardium [21–23]. In these studies, selective pressure-regulated retroinfusion into a coronary vein (AIV) draining the LAD-area was performed during occlusion of the (LAD), an approach also used in patients [24]. Using this application method, we now studied regional pharmacological therapy with GSH and/or cariporide at the end of ischemia and during the onset of reperfusion (a protocol feasible in patients, Kupatt and Boekstegers, unpublished results). Moreover, since GSH and cariporide are widely accepted as acting on myocyte compartment, we studied whether additional protection of the endothelial compartment—by a CD18 antibody inhibiting endothelium–leukocyte interaction—would provide enhanced cardioprotection, or if an overlap of the therapeutic targets of GSH/cariporide treatment and CD18 antibody would exist.
2 Materials and methods
2.1 Materials
Cariporide was a gift of Aventis (Frankfurt, Germany), GSH was purchased from Roche (Milano, Italy). All further chemicals were from Sigma (Deisenhofen, Germany), if not stated otherwise. The CD18 antibody IB4 was raised from a hybridoma cell line from ATCC (New York, USA). Animals were purchased from Oberschleissheim, Germany.
2.2 Cell culture experiments
Rat neonatal cardiomyocytes were prepared as previously published [25]. Thereafter, cells were plated in 12-well plates and subjected to GSH (10 mg/ml) and/or cariporide (100 μg/ml) incubation. Myocytes were challenged by oxidative stress with two protocols: either, after pretreatment with GSH (10 mg/ml, 1 h) and gentle washing, cells were subjected to normoxia (9 h) or hypoxia (8 h) and reoxygenation (1 h 95% O2 and 5% CO2) (Fig. 1A,B). Alternatively, cells were exposed to H2O2 (0.3 mmol/l, 12 h) in the presence of GSH (10 mg/ml) with or without cariporide (100 μg/ml) (Fig. 1C,D). A score was established to semiquantitatively evaluate myocyte function (0=no contracting cell, 1=singular contracting cells, 2=majority of cells contracting at reduced rate, 3=homogenous contraction at rate of normoxic control cells). Viability of cells was assessed by Trypan blue exclusion, whereas Trypan blue-positive cells were counted as dead.
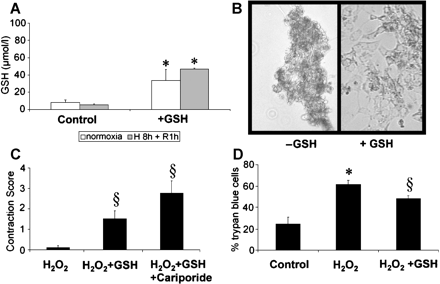
(A) GSH-content of isolated rat neonatal cardiomyocytes (NRCMs) without or with GSH treatment for 60 min (10 mg/ml) and subsequent normoxia (9 h) or hypoxia (8 h) and reoxygenation (1 h). GSH levels of the cell pellets were increased after GSH-exposure. (B) Myocyte cell integrity after 8 h hypoxia and 1 h of reoxygenation (H/R) in untreated NRCMs (left panel) in comparison to GSH-preincubation (right panel). Representative example of three independent cell preparations after Trypan blue staining. (C) NRCMs incubated with GSH (10 mg/ml) with or without cariporide (100 μg/ml) were exposed to H2O2 (0.3 mM) for 12 h and the contractility was scored (cf. Materials and methods). Quantification of four independent cell preparations. (D) Viability of NRCMs (Trypan blue exclusion) after identical H2O2 stimulation without or with GSH exposure. (*=p<0.05 vs. control, §=p<0.05 vs. H2O2).
2.3 Animal experimental protocol
The care of the animals and all experimental procedures conform with the German and NIH animal legislation guidelines and were approved by the local animal care and use committee (AZ 2531-7/01). All pig experiments were conducted at the Institute for Surgical Research of the University of Munich. German farm pigs (n=5 per experimental group) were anesthetized and instrumented as described previously [26]. The external jugular vein and the carotid artery of the right side were cannulated and appropriate sheaths (8 F) were placed.
2.4 Ischemia
A balloon was placed in the LAD distal to the bifurcation of the first diagonal branch, and inflated with 4 atm (0.41 MPa). Correct localisation of the coronary occlusion and patency of the first diagonal branch was ensured by injection of contrast agent. At 30 min of ischemia, systemic CD18-antibody IB4 infusion was performed for 15 min (1.5 mg/kg bodyweight), where indicated, thus targeting circulating leukocytes potentially recruited to the postischemic myocardium.
2.5 Retroinfusion
As described in detail previously [27], the retroinfusion catheter was advanced into the anterior interventricular vein (AIV) draining the parenchyma perfused by the LAD. After assessment of the individual systolic occlusion pressure of the venous system, retroinfusion pressure was set 20 mm Hg above the latter. In retrogradely treated groups, at 55 min of ischemia, continuous pressure-regulated retroinfusion of isothermic Tyrode solution (50 ml/min) containing GSH (250 mg/kg) and, where indicated, coinfusion of cariporide (1 mg/kg), was begun. Retroinfusion was interrupted for 30 s for drawing of blood samples at indicated time points (cf. Fig. 2) and otherwise continued for 10 min, overlapping with the restoration of blood flow for 5 min.
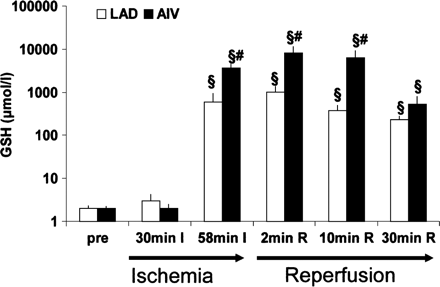
GSH-levels of coronary arterial (LAD) and coronary venous (AIV) blood samples drawn before LAD occlusion (pre), mid-ischemic (30 min I), after the onset of retroinfusion of GSH (58 min I), during retroinfusion and reperfusion (2 min R), and during reperfusion after cessation of retroinfusion (10 min R and 30 min R). As expected, GSH elevation was observed in coronary venous and, to a lesser extent, in coronary artery samples during GSH-retroinfusion. Twenty-five minutes after the end of retroinfusion, coronary venous levels were still >100-fold above pre-ischemic levels (§=p<0.05 vs. pre-ischemic levels, #=p<0.05 vs. time-matched arterial level (n=4).
Blood samples were quickly drawn from coronary ostium and anterior interventricular vein in a random order up to 30 min of reperfusion. Thereafter, venous and arterial catheters and sheaths were withdrawn, the vessels ligated and suture of the muscle and the skin performed. Animals were brought back to the animal facility where they were fed with water and nutrition ad libitum.
At the end of the experiment (day 7 after ischemia), animals were brought back to the OR, anesthetized and instrumented. After hemodynamic measurements, sternotomy was performed and the pericardium was removed. Ultrasonic crystals were placed 1 cm (area at risk) and 4 cm distal to the balloon occlusion site. Each pair was inserted with its axis perpendicular to the LAD. Another pair of crystals was inserted in the Cx perfusion area. Subendocardial segment shortening (SES) was performed under resting heart rate as well as at 120 and 150/min atrial pacing (for 1 min each), as described before [27].
2.6 Infarct size determination
Before arrest of the heart, 20 ml of methylene blue were injected in the left ventricle during external ligation of the LAD at the previous occlusion site, thus staining the non-ischemic area. After heart excision, the infarct size was determined by pressure-controlled injection (80–100 mm Hg) of 15 ml of 10% tetrazolium red into the LAD, staining non-infarcted area at risk intensely red, whereas infarcted tissue remained pale. Thereafter, the left ventricle was cut into slices of 5 mm thickness, which were subjected to digital photography. Image processing of the digital pictures with trace measurements (SigmaScan) provided volume of infarct region, area at risk and total LV area for each slice [28].
2.7 GSH measurements
Blood samples from the coronary artery (LAD) and coronary vein (AIV) were analyzed for GSH content. In addition, tissue samples from a control region (Cx perfusion area), the area at risk and the infarcted area were taken at 1 h of reperfusion (Fig. 3). Total soluble glutathione (GSH+GSSG) was measured in medium, cell pellets, plasma and homogenates from freeze-clamped hearts. Blood samples of 500 μl were collected for determination of GSH and GSSG in plasma. For GSSG analysis, an aliquot (200 μl) of blood was mixed immediately with 200 μl of 10 mM N-ethylmaleimide (NEM) in 100 mM phosphate buffer (pH 6.5) containing 17.5 mM EDTA. The remaining blood was centrifuged at full speed for 1 min. An aliquot (100 μl) of plasma was pipetted into 100 μl sulfosalicylic acid (5%) for determination of total glutathione. To separate GSSG from NEM and NEM-GSH adducts, an aliquot of NEM treated plasma was passed through a Sep-PakC18 cartridge (Waters, Framingham, MA) followed by 1 ml of 100 mM phosphate buffer (pH 7.5). GSSG in the eluates and total glutathione in plasma and acidic homogenates were determined by an enzymatic test as described previously [29]. GSH plasma concentrations were calculated as the difference between total GSH and GSSG.
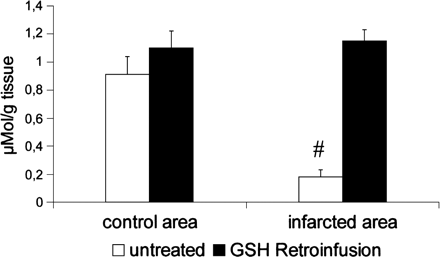
Tissue content of GSH in a non-ischemic control area (Cx perfusion area) and the infarcted area in hearts explanted after 60 min of ischemia and 60 min of reperfusion (n=3 hearts). Whereas infarcted tissue displays a significant loss of GSH compared to non-ischemic RCx-perfusion area, GSH supplementation provided similar GSH levels in the control and infarct area.
2.8 Statistical methods
The results are given as mean±SEM. Statistical analysis was performed using one-way analysis of variance (ANOVA). Whenever a significant effect was obtained with ANOVA, we performed multiple comparison tests between the groups using Student–Newman–Keul's procedure (SPSS statistical program). Differences between groups were considered significant for p<0.05.
3 Results
3.1 GSH-content of rat neonatal myocytes
As depicted in Fig. 1A, GSH content of untreated neonatal rat cardiomyocytes (NRCM) was not significantly different after either normoxia (9 h) or hypoxia (8 h) and reoxygenation (1 h). However, an increased GSH content of the cell pellet was found after exposure to GSH (1 h, 10 mg/ml) before the hypoxia/reoxygenation challenge.
In the given cell culture model, in vitro analysis of the ROS scavenging potential of GSH was performed by exposure to H2O2 (0.3 mmol/l). As depicted in Fig. 1C, regular contractions of the myocytes almost completely ceased 12 h after exposure to H2O2. However, concomitant presence of GSH preserved residual contractile function, a phenomenon further increased by co-application of cariporide (100 μg/ml). Consistently, after H2O2 exposure, the proportion of lethally injured (Trypan blue-positive) NRCMs was limited by GSH (Fig. 1D).
3.2 GSH-content of plasma and tissue samples in vivo
Coronary venous and arterial GSH levels were assessed during the course of GSH retroinfusion. Compared to control levels (2–3 μM), retroinfusion of GSH instantaneously increased coronary venous levels >1000-fold (cf. Fig. 2). This level was sustained up to 5 min after cessation of GSH-retroinfusion (=10 min reperfusion). Concomitant coronary arterial levels of GSH were increased to a significantly lower level than coronary venous levels (cf. Fig. 2). Of note, 25 min after cessation of retroinfusion (=30 min reperfusion) coronary venous GSH levels, declining from peak values, were still >150-fold elevated above untreated samples, however, not differing from arterial GSH levels.
Tissue samples of hearts (Fig. 3) reperfused for 1 h after ischemia revealed that GSH content in the infarcted region of untreated hearts was substantially reduced in comparison to GSH content of the non-ischemic RCx perfusion area (0.18±0.05 vs. 0.91±0.13 μmol/g tissue). However, after GSH retroinfusion, we detected no significant GSH deficiency in the infarct region in comparison to the normoxic control region.
3.3 Infarct size
At day 7 of reperfusion, infarct size of control hearts was 51±5% of the area at risk, a proportion not significantly changed by systemic infusion of GSH/cariporide (Fig. 4A). GSH retroinfusion tended to decrease infarct size, however, missing statistical significance (41±3%) similar to cariporide retroinfusion alone (42±6%). The combination of GSH and cariporide retroinfusion significantly reduced infarct size to 29±3% (p<0.02).
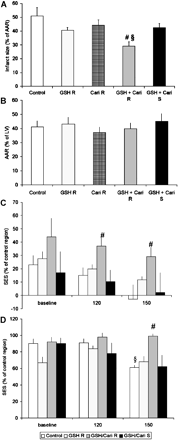
(A) Infarct size was assessed as the ratio of the infarcted area to the area at risk (AAR). Compared to untreated controls, systemic application of GSH and cariporide (GSH Cari S) did not significantly reduce the infarcted area, similarly to retroinfusion of GSH (GSH R) or cariporide (Cari R) alone. In contrast, combined retroinfusion of GSH and cariporide (GSH Cari R) substantially decreased the infarct extension. (B) AAR as of the total LV revealed no difference in infarct size in all groups. (C) Subendocardial segment shortening (SES) in the infarcted area (percentage of the RCx perfusion area) was reduced in untreated control hearts, and was unaltered by GSH retroinfusion alone (GSH R) or systemic GSH/cariporide infusion (GSH Cari S), whereas GSH/cariporide retroinfusion (GSH Cari R) improved SES in the infarcted region at 120 and 150/min pacing rate. (D) SES in the area at risk of the same experimental groups. At 150/min, untreated hearts displayed a loss of SES (compared to baseline), whereas GSH/cariporide retroinfusion preserved regional myocardial function. (n=5; #=p<0.05 vs. control hearts, §=p<0.05 vs. baseline).
3.4 Regional myocardial function
To assess myocardial function in the region exposed to ischemia and reperfusion, sonomicrometry was performed in a standardized manner in the infarcted and non-infarcted area at risk as well as in the non-ischemic CX perfusion area. As depicted in Fig. 4C, subendocardial segment shortening (SES) of the infarcted area was severely reduced to <50% of the non-ischemic area in all groups under resting conditions. In hearts without treatment or with GSH retroinfusion alone or systemic GSH/cariporide application, SES tended to further decline at higher heart rates (pacing at 120 and 150/min), whereas retroinfusion of GSH and cariporide combined preserved SES in the infarcted region under these conditions.
In the area at risk, regional myocardial function was well-preserved at rest (Fig. 4D). As expected, pacing revealed a decreased functional reserve (57±15% at 150/min) in hearts subjected to ischemia and reperfusion without treatment. No loss of regional function was observed when GSH/cariporide retroinfusion was applied.
3.5 Impact of CD18 blockade (IB4)
Since GSH and cariporide are known protectors of the myocyte compartment, we assessed the therapeutic potential of additional leukocyte inhibition by application of a CD18 antibody. We therefore infused IB4 at 30 min of ischemia in a dose of 1.5 mg/kg bodyweight. Time course studies revealed that at the given dose IB4 blocked >99% of the MAC-1, with 50% inhibition present at 4 h (data not shown). Thus, IB4 application appeared suitable for inhibition of early overwhelming inflammation after ischemia and reperfusion.
In fact, IB4 displayed a distinct reduction of infarct size at day 7 (38±4%), although no significant improvement of regional myocardial function occurred. Of note, combining IB4 infusion with GSH/cariporide retroinfusion did not extend the effect of either IB4 or GSH/cariporide treatment (Figs. 4 and 5) significantly.
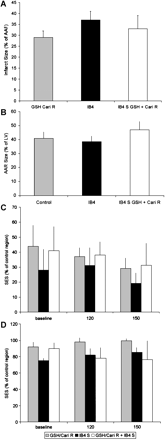
(A) Infarct size after GSH/cariporide retroinfusion (GSH Cari R) was not significantly differing from infarct size after anti CD18 antibody IB4 infusion. No additional effect was found after combination of both treatments. (B) No difference was found in AAR to LV ratio of the three groups. (C,D) SES of the infarct area (C) or the AAR (D) displayed unaltered levels after either GSH/cariporide retroinfusion or IB4 infusion or both.
4 Discussion
In the present study, we used a pig model of 60 min LAD occlusion and 7-day reperfusion to investigate the cardioprotective effect of regional application of GSH and cariporide into the ischemic myocardium. We took advantage of a novel percutaneous retrograde delivery system targeting GSH and cariporide to the ischemic myocardium. Blood sample analysis revealed a rapid increase of GSH in the coronary venous blood, which exceeded coronary artery levels more than 6-fold, indicating effective regional supply of the pharmacologic agents. With this application strategy, we could demonstrate for the first time that the combination of GSH and cariporide retroinfusion decreased infarct size and improved regional function of the area at risk, although the application-to-reperfusion interval comprised a short interval (5 min). Of note, retroinfusion of GSH/cariporide combined provided postischemic cardioprotection which was not achieved by one agent alone nor via systemic application of both (Fig. 4A–C). The additional application of the CD18 antibody IB4 did not induce a further protective effect.
During postischemic reperfusion, one major sources of reactive oxygen species (ROS) are mitochondrial enzymes of the myocytes [30]. ROS might injure myocytes, e.g. by mitochondrial cytochrome c release [31–33], associated with myocyte apoptosis. Myocyte protection against oxidative stress was achieved by GSH supplementation in rat neonatal myocytes challenged by H2O2(Fig. 1C,D) or hypoxia/reoxygenation (Fig. 1A,B). Here, cell survival as well as cell morphology and function (contraction) was preserved to a larger extent after GSH exposure. Of note, GSH was capable of reducing cellular detriment induced by hypoxia/reoxygenation although GSH incubation (1 h) was interrupted with the onset of hypoxia (Fig. 1A,B). Similar results have been obtained with adult cardiomyocytes in cell culture [34]. In vivo, retroinfusion of GSH was able to compensate for the early loss of GSH in the infarcted area (at 1 h of reperfusion), similar to results of other in vivo studies in pigs [14,35]. However, GSH application alone did not suffice to significantly reduce infarct size in our study. This observation might point to the depth of ischemia, since cardioprotection by 5 mM GSH after 20 and 30 min of hypoxia was lost after 50 min of hypoxia in isolated rat hearts [13]. On the other hand, the cardioprotective effect of endogenous GSH shown by depletion strategies in rats [36], dogs [37] and pigs [14] was exclusively achieved after pre-ischemic GSH-depletion in each study. This therapeutic time frame is reflected by the cardioprotective effect of GSH after supplementation throughout ischemia in vivo [35]. In the present study, late ischemic GSH retroinfusion, although rapidly increasing local GSH plasma levels (Fig. 2) as well as GSH tissue content (Fig. 3), did not exert significant cardioprotection, despite a trend towards smaller infarct size (Fig. 4A). This lack of therapeutic efficacy in vivo cannot be attributed to inefficient distribution of the agent retroinfused into the ischemic region (cf. Fig. 2), as opposed to systemic application. More likely, the depth of 1 h ischemia (cf. Ref. [13]) as well as detriment occurring early during ischemia might have prevented a more pronounced effect of GSH retroinfusion.
Unfortunately, clinical therapy of acute myocardial infarction most often faces an ischemic time of varying length, which has to be limited by rapid reperfusion. This scenario excludes pharmacological treatment early during limited ischemia, as obviously required for GSH to be effective. Since a further increase of GSH did not appear practical, we sought to extend cardioprotection by combination of GSH and cariporide. The different mechanisms of action of cariporide, e.g. prevention of calcium overload, suggested that an overlap with the ROS-scavenger GSH might be limited, although both agents are capable of protecting myocytes [34,38]. In fact, despite a lack of significant cardioprotection achieved by retroinfusion of cariporide alone, the combination with GSH retroinfusion provided a substantial reduction of infarct size and regional myocardial dysfunction (Fig. 4). Noteworthy, the cardioprotection was achieved by retroinfusion into the ischemic myocardium not earlier than 5 min before reopening of the occluded coronary artery. Rather than covering the ischemic period, our regional treatment approach was targeted at the onset of reperfusion.
As a common denominator, GSH and cariporide are widely viewed as preferentially protecting the myocyte compartment. To test if GSH/cariporide application leaves the endothelium unprotected against postischemic dysfunction, we applied a CD18 antibody (IB4), which was able to reduce infarct size (Fig. 4) by virtue of inhibiting leukocyte adhesion on the endothelium, as demonstrated before [22]. Co-application of GSH/cariporide and IB4 did not further limit reperfusion injury in terms of infarct size and regional function. This finding indicates that endothelial protection is also a feature of GSH/cariporide treatment, as suggested by recent studies [39–41]. E.G. the antiinflammatory properties of resting endothelium, such as moderate nitric oxide release [4] or strictly limited adhesion molecule expression [42] are typically lost after ROS exposure. Both, however, are maintained by GSH [41,43] and cariporide [44], enabling less PMN accumulation in the infarct area of the GSH/cariporide retroinfused hearts (data not shown). Moreover, cariporide appears to decrease PMN activation after stimulation with ROS [45].
In summary, the present study demonstrates that selective retroinfusion of GSH and cariporide provides an effective and timely approach to limiting postischemic myocardial reperfusion injury. This approach proved to be superior to therapy with either agent alone or systemic venous application of both. Moreover, an overlap with endothelial protection is suggested by the lack of additional CD18 blockade. These results have prompted the initiation of a clinical study in patients with acute myocardial infarction, who are currently being treated with GSH/cariporide retroinfusion 5 min before the onset of reperfusion, as outlined above.
Acknowledgements
This study was supported by a grant from the Deutsche Forschungsgemeinschaft. The expert assistance of Elisabeth Ronft, Susanne Helbig and Ingrid Liss is gratefully acknowledged.
References
Author notes
Both authors contributed equally to this manuscript.
Time for primary review 29 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}