





Abstract
On the basis of its ability to inhibit fibrosis, pirfenidone has drawn the attention as an intriguing candidate for treating cardiac disease. However, its precise electrophysiological effects have yet to be elucidated. Here, we have investigated its potential to modulate ion channels.
Adult rat cardiac myocytes were investigated using whole-cell patch-clamp, western-blot and qRT-PCR techniques. Pirfenidone increased the density of L-type Ca2+ current (ICaL, 50–100%), without significantly altering Na+, K+, or T-type Ca2+ currents. The effect was dose-dependent, with an EC50 of 2.8 µM. Its onset was slow, with a lag period larger than 1 h and time to maximum of 24–48 h. Concomitant changes were observed in the voltage-dependent activation of ICaL (−5 mV shift in both V1/2 and k). In contrast, the following properties of ICaL remained normal: steady-state inactivation, CaV1.2 levels (mRNA and protein), and intramembrane charge movement. Indeed, the conductance-to-charge ratio, or Gmax/Qmax, was increased by 80%. The effect on ICaL was mimicked by an inhibitor of nitric oxide (NO) synthase (NOS), and attenuated by both cyclic adenosine monophosphate (cAMP) and cAMP-dependent protein kinase (PKA) inhibitors. Conversely, cytokines, reactive oxygen species, and Ca2+ were all ruled out as possible intermediaries. Additional experiments suggest that pirfenidone increases action potential duration by ∼50%.
Pirfenidone augments ICaL, not through higher expression of L-type channels, but through promoting their Ca2+-conducting activity. A possible inhibition of NOS expression is likely involved, with subsequent reduced NO production and stimulated cAMP/PKA signalling. These findings may be relevant to the cardioprotective effect of pirfenidone.
1. Introduction
The compound pirfenidone (5-methyl-phenyl-2-(1H)-pyridone) is an anti-fibrotic agent of broad spectrum that has been approved for the treatment of idiopathic pulmonary fibrosis. Its anti-fibrotic effects can be largely explained by a concomitant inhibition of the expression of transforming growth factor-β1 (TGF-β1, the most prominent pro-fibrotic cytokine). Pirfenidone also inhibits the expression of the pro-inflammatory cytokine tumour necrosis factor-α (TNF-α), and is thus considered a potent anti-inflammatory (reviewed in refs1–3). In addition to its anti-fibrotic and anti-inflammatory effects, pirfenidone also inhibits the expression of nitric oxide (NO) synthase (NOS); and accordingly, it also reduces the levels of NO.4–6 While these effects occur over the time frame of several hours or days, in vitro experiments indicate that pirfenidone also acts rapidly, within seconds, as a scavenger of reactive oxygen species (ROS).7,8
One may expect that pirfenidone results in cardiac beneficial effects, as fibrosis hinders the function of the heart. As a matter of fact, this compound attenuates cardiac stiffness9,10 and also reduces the risks of atrial fibrillation (AF),11 myocardial infarction,12 and ventricular tachycardia.12 Although these observations have been correlated with a concomitant reduction of fibrosis, evidence exist that pirfenidone can also improve cardiac contractility even in the absence of fibrosis and stiffness.13 Overall, these studies suggest that pirfenidone represents an intriguing candidate to treat cardiac disease. However, there is no information about its possible impact on ion channels. Here, we show that in both atrial and ventricular myocytes, a chronic pirfenidone treatment drastically enhances the activity of L-type Ca2+ channels (L-channels). We also provide insights about both the underlying molecular mechanisms and the signalling pathways that may be involved. Our findings may represent a parallel mechanism by which, in addition to reduced fibrosis, pirfenidone exerts its cardioprotective effects.
2. Methods
A more complete version of the Methods can be found as Supplementary material online.
2.1 Primary cultures of cardiac myocytes
This protocol was designed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011), NRC 2011, complies with the Mexican Official Norm NOM-062-ZOO-1999, and was approved by the Institutional Animal Care and Use Committee (IACUC—Cinvestav, 0259-05). Briefly, male Wistar rats (weighting ∼250 g) were heparinized (1–1.25 U/g), and a mixture of ketamine–xylazine was applied (100:10 mg/kg of body weight, ip). Once the animal was anaesthetized, as judged by absence of spontaneous movements and reflex responses, the heart was excised and perfused retrogradely using a homemade Langendorff system (set at 37°C). Following an enzymatic digestion, the tissue of interest (ventricles, left atria, or right atria) was dissected and mechanically triturated. Cells were plated in Petri dishes containing standard culture medium (control condition). Pirfenidone and other compounds were added from concentrated stocks to the standard culture medium, to result in the final concentrations indicated.
2.2 Current-clamp experiments
Action potentials (APs) were recorded under current-clamp conditions, using a slightly modified method originally described by Ferreiro et al.14 Briefly, the whole-cell configuration was obtained under voltage clamp, the amplifier was switched to the fast current-clamp mode, and Vm was set to ∼−80 mV. APs were induced at 1 Hz by the application of brief (0.5 ms) pulses of depolarizing current, in the presence of Tyrode solution.
2.3 Voltage-clamp experiments
Voltage-clamp experiments were performed using procedures described previously.15,16 Basically, the cells were transferred from the incubator to a recording chamber, mounted into an inverted microscope, and subjected to voltage-clamp experiments, using the whole-cell patch-clamp technique. In certain figures, the results are shown normalized because it facilitates visual comparison of pirfenidone effects. The only factor needed to decode these data is the absolute mean of controls, which is given in figure legends.
2.4 Recording solutions
Three different sets of recording solutions were used for voltage-clamp experiments (with a pH of 7.30):
(i) To investigate Na+ and K+ currents, the external and internal recording solutions consisted, respectively, of (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 0.5 CdCl2, and 10 glucose; and 115 KCl, 30 NaCl, 1 CaCl2, 10 EGTA, 10 HEPES, 2 MgCl2, 2 Na2ATP, 0.05 Na2GTP, and 5 glucose.
(ii) Ca2+ currents were investigated using the following external and internal recording solutions, respectively (in mM): 130 methanesulfonic acid, 10 HEPES, 2 or 5 CaCl2, 2 MgSO4, 115 TEA-OH, 5 4-aminopiridine, 1 9-anthracene carboxylic acid, 2 2,3-butanedione monoxime, and 0.06 TTX; and 140 Cs-Asp, 10 Cs-EGTA, 2 CaCl2, 5 ATP-Mg, 10 CsCl, 5 glucose, 0.05 GTP-Tris, and 10 HEPES. These solutions were also used in the experiments of Figure 2E. When indicated, ICaL was recorded with the aforementioned internal solution but supplemented with either cyclic adenosine monophosphate (cAMP, 100 µM) or a cAMP-dependent protein kinase (PKA) inhibitor (PKI, 10 µM). In one experiment, the membrane-permeable PKA inhibitor H89 was added to this extracellular solution.
(iii) Finally, the voltage dependence of intramembrane charge movement (QON) was investigated with the same solutions as Ca2+ currents (ii), except that the external solution contained 2 mM CaCl2, 3 mM CdCl2, 0.1 mM LaCl3, and 0.003 TTX.
2.5 Quantitative real-time PCR assays
The CaV1.2 mRNA levels were assessed by qRT-PCR, using specific primers and procedures similar to those described previously,15 except that here we used the SYBR Green method on an Applied Biosystems 7500 Real-Time PCR system. The targeted sequence includes that encoded by exon 1a, which represents the main isoform reported for cardiac myocytes.
2.6 Western blot
The expression levels of CaV1.2 protein were assessed by western-blot analysis. Basically, myocytes from pooled atria and ventricles were cultured 1 day under either control conditions or pirfenidone, washed twice on ice-cold PBS, and then exposed to a hypotonic solution (10 mM HEPES, 0.5 mM MgCl2) for 15 min on ice. Myocytes were then lysated by adding an equal volume of 2× Laemmli sample buffer. Protein samples (25 µL) were subject to SDS–PAGE, transferred onto PDVF membranes, and subject to western blot as described before.16 Monoclonal antibody anti-Cav1.2, clone L57/46, was obtained from the UC Davis/NIH NeuroMab Facility (Davis, CA, USA), and secondary HRP-conjugated anti-mouse antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Band analysis was performed with ImageJ software. Band intensity was normalized to the sum of all bands in protein samples stained with Coomassie Blue.
2.7 Statistical analysis
All results are presented as media ± SE; unless specified, significant differences were determined at the P < 0.05 level, using Student’s' unpaired t-tests (two-tailed).
3. Results
3.1 Pirfenidone stimulates L-type but not T-type Ca2+ channels
Figure 1A shows low- and high-threshold Ca2+ currents that are in good agreement with the well-known hallmarks of T- (ICaT) and L-type (ICaL) Ca2+ currents, respectively, from adult rat atrial myocytes.17 Interestingly, in both left and right atrial myocytes, pirfenidone significantly increased the density of ICaL, but did not alter ICaT (Figure 1A–C). Thus, pirfenidone selectively stimulates the activity of L-channels. This notion was further tested by recording high-threshold Ca2+ currents in the presence of the dihydropyridine (DHP) antagonist nimodipine. Basically, the corresponding results indicate that 3 µM of nimodipine totally reverts the increase in Ca2+ current (see Supplementary material online, Figure S1A). In fact, pirfenidone significantly increased the DHP-sensitive current by ∼2.3-fold (see Supplementary material online, Figure S1B). From this, one can conclude that a chronic treatment (1–2 days) with pirfenidone selectively targets the cardiac L-channel.
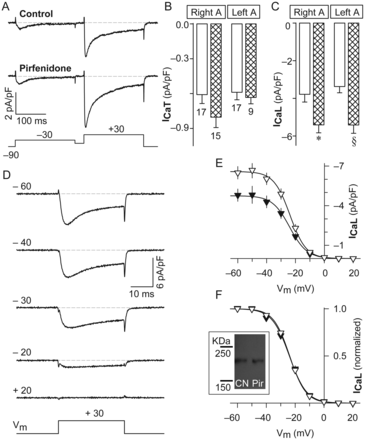
Selective effects on L-type vs. T-type channels. (A) Representative T- (−30 mV) and L-type (+30 mV) Ca2+ currents recorded from right atrial myocytes that were kept in culture in the absence or the presence of pirfenidone (the interpulse potential was −50 mV). (B and C) Average values of T- (ICaT) and L-type (ICaL) Ca2+ currents that were recorded as in (A) from either control cells (white bars) or pirfenidone-treated cells (hatched bars). Results are from myocytes obtained from left ( left A) and right ( right A) atria, as indicated. The number of experiments is also being indicated in (B). The symbols indicate significant differences compared with respective controls (*P < 0.01 and §P < 0.001). (D) Examples of current traces (obtained from a control cell) that were analysed to investigate the steady-state voltage dependence of inactivation of L-channels. (E and F) Absolute (E) and normalized (F) values of ICaL that were acquired as in (D). These results were obtained from both left and right atria, and represent the average from a total of 22 control (closed symbols) and 20 pirfenidone-treated (open symbols) myocytes. Inset shows a representative immunoblot for CaV1.2 in control (CN) and pirfenidone-treated (Pir) cells. The average values of CaV1.2 protein obtained from CN and Pir were (in arbitrary units): 7.0 ± 2.2 and 8.9 ± 3.5, respectively (P = 0.7, n = 3).
3.2 The effect is not due to changes in CaV1.2 inactivation or expression
The effect in ICaL could be potentially explained by assuming that in control cells there is a higher fraction of inactivated channels. Thus, we aimed to investigate the steady-state voltage dependence of inactivation of L-channels. Interestingly, however, in both groups of cells, the fraction of inactivated channels was practically identical at all voltages (Figure 1D–F). Moreover, apart from a ∼40% increase in the maximal density if ICaL (Imax), other parameters describing the voltage dependence of inactivation were not significantly altered (see Supplementary material online, Table S1). Thus, pirfenidone does not act by reducing the fraction of inactivated channels.
On the other hand, if the increase in ICaL was due to a higher expression of the principal subunit of L-channels (CaV1.2), then the corresponding levels of mRNA could be higher in treated cells than in controls. However, the CaV1.2 mRNA levels were practically identical in both control and pirfenidone-treated cells (1.0 ± 0.4 and 1.0 ± 0.3, respectively; n = 6). Moreover, immunoblot analysis indicated that pirfenidone does not significantly alter the levels of CaV1.2 protein (inset of Figure 1; see also Supplementary material online, Figure S6). Thus, these data indicate that pirfenidone increases ICaL without significantly altering expression levels of CaV1.2 (mRNA and protein).
3.3 Pirfenidone increases the current-to-charge ratio
We next investigated the possibility that pirfenidone might be increasing the number of L-channels at the plasma membrane. This was done by measuring immobilization-resistant charge movements. However, as can be seen in Figure 2A, the families of gating currents that were obtained from both control and pirfenidone-treated cells were very similar. As a matter of fact, both the absolute values of charge movement and the corresponding voltage dependence were also practically identical among experimental groups (Figure 2B and C, and Q–V data of Table 1). Thus, in keeping with the absence of changes in CaV1.2 (inset of Figure 1F), pirfenidone treatment does not seem to alter the number of L-channels in the plasma membrane.
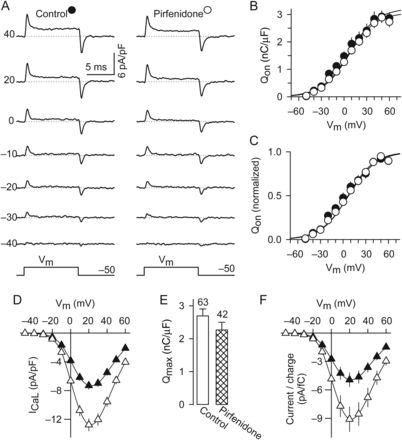
Increases in the current-to-charge ratio. (A) Examples of immobilization-resistant charge movements that were recorded from control and pirfenidone-treated cells. (B and C) Absolute (B) and normalized (C) values of charge movements that were recorded at the onset of depolarization (QON), as illustrated in (A). The parameters obtained by fitting the experimental data shown in (B) by Eq. (4) are given in Table 1 (Q–V data). (D–F) I–V curves (D), Qmax (E), and current-to-charge ratios (F) that were simultaneously recorded from a total of 63 control and 42 pirfenidone-treated cells. Qmax (E) was estimated from current traces with zero ionic current and at the apparent reversal potential, as described in McDonough et al.38 The resulting average values of Gmax/Qmax, as well as the other Boltzmann parameters describing the I–V curves are given in Table 1 (G–V data).
Parameters of fitted I–V and Q–V curves
| G–V data | Q–V data | |||||||
|---|---|---|---|---|---|---|---|---|
| Gmax (nS/nF) | VG,1/2 (mV) | KG (mV) | Vrev (mV) | Qmax (nC/µF) | VQ,1/2 (mV) | KQ (mV) | Gmax/Qmax (nS/pC) | |
| Control | 181 ± 10 (62) | 8.7 ± 0.6 (62) | 8.7 ± 0.1 (62) | 71 ± 1.0 (62) | 3.2 ± 0.2 (16) | 7.3 ± 3.4 (16) | 18.5 ± 1.5 (16) | 115 ± 16 (62) |
| Pirfenidone | 290 ± 18* (40) | 7.3 ± 0.4* (40) | 8.2 ± 0.1* (40) | 73 ± 0.5 (40) | 3.1 ± 0.2 (21) | 6.6 ± 2.3 (21) | 17.6 ± 0.8 (21) | 203 ± 28* (40) |
| G–V data | Q–V data | |||||||
|---|---|---|---|---|---|---|---|---|
| Gmax (nS/nF) | VG,1/2 (mV) | KG (mV) | Vrev (mV) | Qmax (nC/µF) | VQ,1/2 (mV) | KQ (mV) | Gmax/Qmax (nS/pC) | |
| Control | 181 ± 10 (62) | 8.7 ± 0.6 (62) | 8.7 ± 0.1 (62) | 71 ± 1.0 (62) | 3.2 ± 0.2 (16) | 7.3 ± 3.4 (16) | 18.5 ± 1.5 (16) | 115 ± 16 (62) |
| Pirfenidone | 290 ± 18* (40) | 7.3 ± 0.4* (40) | 8.2 ± 0.1* (40) | 73 ± 0.5 (40) | 3.1 ± 0.2 (21) | 6.6 ± 2.3 (21) | 17.6 ± 0.8 (21) | 203 ± 28* (40) |
*Compared with control, P < 0.05.
Parameters of fitted I–V and Q–V curves
| G–V data | Q–V data | |||||||
|---|---|---|---|---|---|---|---|---|
| Gmax (nS/nF) | VG,1/2 (mV) | KG (mV) | Vrev (mV) | Qmax (nC/µF) | VQ,1/2 (mV) | KQ (mV) | Gmax/Qmax (nS/pC) | |
| Control | 181 ± 10 (62) | 8.7 ± 0.6 (62) | 8.7 ± 0.1 (62) | 71 ± 1.0 (62) | 3.2 ± 0.2 (16) | 7.3 ± 3.4 (16) | 18.5 ± 1.5 (16) | 115 ± 16 (62) |
| Pirfenidone | 290 ± 18* (40) | 7.3 ± 0.4* (40) | 8.2 ± 0.1* (40) | 73 ± 0.5 (40) | 3.1 ± 0.2 (21) | 6.6 ± 2.3 (21) | 17.6 ± 0.8 (21) | 203 ± 28* (40) |
| G–V data | Q–V data | |||||||
|---|---|---|---|---|---|---|---|---|
| Gmax (nS/nF) | VG,1/2 (mV) | KG (mV) | Vrev (mV) | Qmax (nC/µF) | VQ,1/2 (mV) | KQ (mV) | Gmax/Qmax (nS/pC) | |
| Control | 181 ± 10 (62) | 8.7 ± 0.6 (62) | 8.7 ± 0.1 (62) | 71 ± 1.0 (62) | 3.2 ± 0.2 (16) | 7.3 ± 3.4 (16) | 18.5 ± 1.5 (16) | 115 ± 16 (62) |
| Pirfenidone | 290 ± 18* (40) | 7.3 ± 0.4* (40) | 8.2 ± 0.1* (40) | 73 ± 0.5 (40) | 3.1 ± 0.2 (21) | 6.6 ± 2.3 (21) | 17.6 ± 0.8 (21) | 203 ± 28* (40) |
*Compared with control, P < 0.05.
To further test the hypothesis that pirfenidone increases ICaL without changing the number of channels, we measured for each particular myocyte the values of Gmax and Qmax. This allowed us to make a statistical comparison of the Gmax/Qmax ratio (Table 1), as well as to normalize the peak values of ICaL (Figure 2D) by its corresponding Qmax value (Figure 2E) for each myocyte. By these means, we were able to obtain the current-to-charge ratios that are shown in Figure 2F. Basically, these data indicate that, in parallel measurements, pirfenidone treatment increases Gmax (Figure 2D and Table 1) without significantly altering Qmax (Figure 2E). As a consequence, it significantly increases both the current-to-charge ratio (Figure 2F) and the average values of Gmax/Qmax (Table 1). Taken together, the results shown in Figures 1 and 2 provide compelling evidence that pirfenidone does not alter the cell surface expression of L-channels, but enhances the amount of calcium ions that move across the plasma membrane per unit of gating charge moved.
3.4 Effects in voltage dependence of activation
In addition to increasing the current-to-charge ratio of L-channels (Figure 2 and Table 1), pirfenidone also altered their voltage dependence of activation. The following evidence provides support for this view. First, the I–V curves shown in Figure 2D indicate minor but significant negative shifts in the Boltzmann parameters VG,1/2 and kG (see G–V data in Table 1). Second, though these changes were rather small, they were also detected in ventricular myocytes (see Supplementary material online, Figure S2). More precisely, in these cells, pirfenidone also not only increased Gmax, but reproduced the negative shifts in both VG,1/2 and kG (see Supplementary material online, Figure S2). Thus, a shift in channel activation towards negative potentials and a reduction in the slope factor were systematically observed. These data suggest that pirfenidone produces a moderate but significant increase in voltage sensitivity of L-channels.
3.5 Timing and dose-dependence of L-channel modulation
The effects on ICaL are rather persistent as pirfenidone had been washed out 10–60 min before the recordings. However, as described below, their onset requires continuous presence of the drug.
As can be seen in Figure 3A, in control atrial myocytes, by 18 h of culture, the basal levels of ICaL spontaneously decreased to a 50%, and remained relatively low until at least 24 h later. In contrast, in atrial myocytes exposed to pirfenidone; throughout the entire period of culture (44 h), ICaL remained constant and similar to the initial 100%. Thus, pirfenidone clearly stabilized ICaL to a value that was similar to that observed in freshly isolated atrial myocytes (Figure 3A—atria).
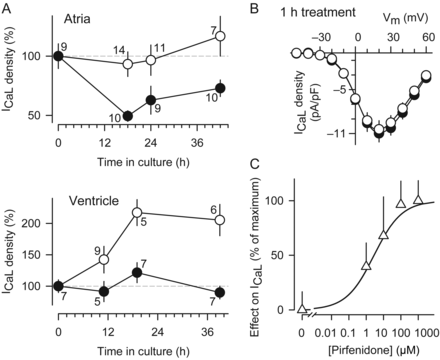
Time course and dose–response of ICaL potentiation. (A) Changes in ICaL as a function of time in culture in either the absence (closed symbols) or the presence (open symbols) of pirfenidone. The absolute value of ICaL in freshly isolated myocytes (i.e. time 0) was −6.7 pA/pF (atria) and −6.8 pA/pF (ventricle). (B) I–V curves obtained from atrial myocytes that were kept in culture 1 day under control conditions, and then either exposed (open symbols, n = 7) or unexposed (closed symbols, n = 7) to pirfenidone during 1 h. (C) Percentage of stimulatory effect on ICaL at different concentrations of pirfenidone. The results were normalized with respect to the maximal increase in current density (∼80%). The treatment with pirfenidone lasted 1–2 days.
In ventricular myocytes, a chronic exposure to pirfenidone also up-regulates the activity of L-channels (see Supplementary material online, Figure S2). The results of Figure 3A extend this observation. It can be seen that, under control conditions, the basal levels of ICaL did not significantly change, at least for the investigated time frame of ∼40 h. In contrast, after adding pirfenidone to the culture medium, the density of ICaL was raised above these levels (Figure 3A—ventricle).
It can be concluded from the results shown in Figure 3A that the effect of pirfenidone on ICaL becomes noticeable after 10 h, is fully established by 18 h, and remains for at least 42 h. More importantly, as shown in Figure 3B, a short-term incubation (of 60 min) of atrial myocytes with pirfenidone did not significantly alter either the density or voltage dependence of ICaL. Thus, pirfenidone evidently modulates the activity of L-channels through a long-term and persistent mechanism, which, surprisingly, does not involve changes in the levels of CaV1.2 mRNA, protein, or charge movement (Figure 1F, its description, and Figure 2).
A dose–response curve is also being shown in Figure 3C. As can be seen, the stimulation of L-channels by pirfenidone is dose-dependent, with an EC50 of 2.8 µM and Hill coefficient of 0.5.
It is well known that cardiac myocytes undergo changes in culture. Accordingly, in our experiments, the borders of ventricular myocytes become rounded, but the cell surface remained striated. In fact, as can be seen in Supplementary material online,Supplementary Data, the average values of Cm were almost identical among all experimental conditions. Similarly, the atrial myocytes become rounded, but this did not result in significant alterations in Cm. Additionally, in these cells, the I–V curve of ICaL was rightward shifted (see Supplementary material online, Figure S4). A similar shift has been also observed previously in ventricular myocytes, as a part of the changes associated with their culturing.18,19
3.6 The modulation of L-channels does not involve cross-talk between pirfenidone and TGF-β1
TGF-β1 is an important promoter of both AF and fibrosis.20 Moreover, pirfenidone prevents the development of AF, probably by inhibiting the expression of both TGF-β1 and fibrosis.11 On the other hand, we have shown that TGF-β1 inhibits the expression of L-channels in neonatal rat atrial myocytes.15 Thus, we hypothesized that perhaps TGF-β1 might prevent the stimulatory effect of pirfenidone on L-channels. However, as can be seen in Figure 4A and B, TGF-β1 neither modified the basal levels of ICaL nor attenuated the augmentation of ICaL produced by pirfenidone.
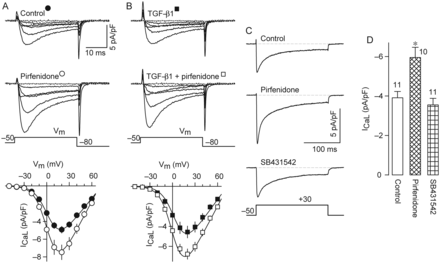
The TGF-β1 signalling is not involved. (A and B) Representative current families of L-channels and corresponding I–V curves (lower plots) that were obtained from myocytes cultured under either control conditions (n = 18) or the presence of 100 nM TGF-β1 (n = 24), pirfenidone (n = 22), or TGF-β1 plus pirfenidone (n = 23). (C) Representative traces of ICaL that were recorded from a control cell and cells cultured in the presence of pirfenidone or a TGF-β1 receptor kinase inhibitor (SB431542, 10 μM). (D) Summary of results obtained from current traces recorded as in (C). *P < 0.005 compared with either control or SB431542.
There is room for argument that there may be tonic inhibition of L-channels by endogenous TGF-β1, and this could explain the failure of exogenous TGF-β1 to reduce ICaL (Figure 4B). If that were the case, then long-term treatment with a specific inhibitor of TGF-β1 receptors should produce an increase in ICaL. However, in contrast to that observed with pirfenidone, the TGF-β1 kinase receptor inhibitor, SB431542, was unable to increase the basal levels of ICaL (Figure 4C and D). Thus, we conclude that pirfenidone stimulates L-channels independently of the TGF-β1 signalling pathway, which at least in neonatal myocytes exerts an opposite effect.15
3.7 The pirfenidone action might be related to inhibition of NOS expression
Pirfenidone has been shown to inhibit the NOS/NO pathway in a time frame that is well-situated to explain the potentiation of ICaL. Specifically, down-regulation of NOS expression and NO production can only be achieved after 3–6 h of treatment.4,5 Thus, involvement of this pathway may account for the 10 h lapse needed to observe the effects on ICaL (Figure 3A). This prompted us to investigate whether chronic treatment of myocytes with a well-known inhibitor of NOS (L-NAME) could be able to mimic the effect of pirfenidone. Interestingly, we found that cells incubated 1–2 days in the presence of L-NAME exhibited a significant increase in the density of ICaL (Figure 5A). Moreover, the effects of pirfenidone and L-NAME were not additive (Figure 5A), suggesting that both compounds act on similar molecular targets. These data along with previous observations4,5 are in keeping with the idea that pirfenidone may stimulate L-channels via lower NOS expression and subsequent reduced NO synthesis.
![Involvement of NO, cAMP, and PKA. (A) Representative traces (left) and corresponding normalized average values (right) of ICaL that were obtained from cells cultured 1–2 days under either control conditions, the presence of pirfenidone, L-NAME (1 mM), or pirfenidone plus L-NAME. The hatched bars indicate experimental conditions involving pirfenidone. The symbols indicate significant differences, compared with control (*P < 0.05 and §P < 0.001). (B) I–V curves for ICaL that were obtained from control (closed symbols) and pirfenidone-treated (open symbols) myocytes, using two different internal recording solutions: standard and supplemented with 100 µM of cAMP. The results were obtained from a total of 34 cells, in either the absence (10 control and eight pirfenidone-treated) or the presence of cAMP (seven control and nine pirfenidone-treated). (C) Extent of the effect of cAMP on ICaL [estimated from (B), *P < 0.005]. (D) Normalized values of ICaL that were obtained from control (open bars) and pirfenidone-treated (hatched bars) cells, in the presence of either standard internal solution (left bars) or internal solution added with 10 µM of PKI 5–24 (right bars). *P < 0.05 compared with respective control. The absolute mean of controls in (A) and (D) was (in pA/pF) −5.5 and −6.1, respectively.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/96/2/10.1093_cvr_cvs248/2/m_cvs24805.gif?Expires=1716382423&Signature=x9WZ6tq7b8kEHcOiDtqb3PQiBQHlmsIgfE6iVbihikdT1Jso53QA2Rolk6lgaPT1mtxx4Wo0KlDwcB8aD7-~x0u5AQiSrxQqPBTKmUCwSgpRqMdqBH2VY~fVJlJomACtunNhPuuMlPlgHH8xU2ibK6kA4etT3rYFaNKn5sN6k2doyEYh7EyQFbm34Q5bRE-R~DnR6jc1ZU6zpVP7O583pQsKxxlKUnPcnzcbOzo9A3lYB6z8KBNYZUq9I-yLZGpK6mG4BLniOEYKLlN8K41vPxXgOhCqDh8Gxv3TyLDwXQzXpZh3Nds48Fjh6LNVYYkWlWSHyAU5BxfuW2MpjYPGFg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Involvement of NO, cAMP, and PKA. (A) Representative traces (left) and corresponding normalized average values (right) of ICaL that were obtained from cells cultured 1–2 days under either control conditions, the presence of pirfenidone, L-NAME (1 mM), or pirfenidone plus L-NAME. The hatched bars indicate experimental conditions involving pirfenidone. The symbols indicate significant differences, compared with control (*P < 0.05 and §P < 0.001). (B) I–V curves for ICaL that were obtained from control (closed symbols) and pirfenidone-treated (open symbols) myocytes, using two different internal recording solutions: standard and supplemented with 100 µM of cAMP. The results were obtained from a total of 34 cells, in either the absence (10 control and eight pirfenidone-treated) or the presence of cAMP (seven control and nine pirfenidone-treated). (C) Extent of the effect of cAMP on ICaL [estimated from (B), *P < 0.005]. (D) Normalized values of ICaL that were obtained from control (open bars) and pirfenidone-treated (hatched bars) cells, in the presence of either standard internal solution (left bars) or internal solution added with 10 µM of PKI 5–24 (right bars). *P < 0.05 compared with respective control. The absolute mean of controls in (A) and (D) was (in pA/pF) −5.5 and −6.1, respectively.
3.8 The cAMP/PKA signalling pathway is involved at the latest step
We next sought to determine whether cAMP and PKA could be involved, since inhibition of this pathway by NO results in down-regulation of ICaL.21 As can be seen in Figure 5B–D, this seems to be the case. First, we reasoned that if the PKA-dependent phosphorylation sites were saturated in L-channels from pirfenidone-treated cells, then the inclusion of cAMP in the patch pipette should attenuate the potentiation of ICaL. Accordingly, while pirfenidone increased ICaL by ∼100% in the absence of cAMP (Figure 5B, left), in the presence of this compound, the increase was only barely resolved (Figure 5B, right). In fact, the extent of cAMP effect on ICaL was significantly smaller in pirfenidone-treated cells compared with controls (Figure 5C). These data indicate that compared with controls, in pirfenidone-treated cells, the L-channels are closer to maximum cAMP-dependent stimulation.
We next wondered whether the potentiation of ICaL could be also attenuated by including a selective inhibitor of PKA (PKI) in the patch pipette. As we have been shown above, in the absence of PKI, ICaL was unsurprisingly increased, in this case by ∼50% (Figure 5D, left bars). However, in the presence of PKI, the increase was smaller and statistically insignificant (Figure 5D, right bars). Moreover, although pirfenidone increased ICaL in the presence of a membrane-permeable PKA inhibitor (H89, added to the extracellular recording solution), the extent of the effect was ∼30% smaller (P < 0.05) compared with a parallel experiment in which H89 was omitted (see Supplementary material online, Figure S5). Thus, we conclude that both cAMP and PKA inhibition significantly attenuate the potentiation of ICaL, suggesting that these molecules might be at least partially involved.
3.9 Absence of effects in K+ and Na+ channels, and impact on APs
We next decided to investigate potential chronic effects on Na+ and K+ channels (Figure 6). IK was divided into three components: IKin, IKto, and IKsus. However, none of them were significantly altered, in either density or voltage dependence (Figure 6A–D). Likewise, the density of INa (accessed at +30 mV) was not statistically different between control and pirfenidone-treated cells (Figure 6E and F). A computerized model of the human atrial AP indicated that a selective increase in the maximal conductance of L-channels should increase the AP duration (APD, data not shown). Hence, we aimed to investigate whether pirfenidone could in fact be able to increase APD. As expected, in pirfenidone-treated cells, APD showed a clear tendency to be longer (Figure 6G).
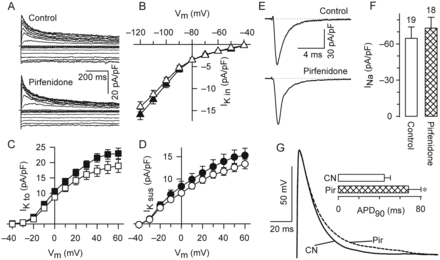
Effects in the atrial AP but not in Na+ and K+ channels. (A) Representative traces of IK. (B–D) Average values of IKin, IKto, and IKsus that were obtained from current traces as in (A). The results were obtained from 53 control (closed symbols) and 40 pirfenidone-treated (open symbols) cells. (E and F) Examples of INa (E) and its corresponding average peak density values (F). (G) Examples of APs that were recorded from control (CN) and pirfenidone-treated (Pir) cells. Inset shows the average values of time to 90% repolarization obtained from a total of 31 myocytes (17 CN and 14 Pir, *P < 0.1).
3.10 Potential participation of other signalling molecules
Pirfenidone is well known for its activity as scavenger of ROS.7,8 Thus, we performed additional experiments to test whether ROS might be involved in the chronic stimulation of L-channels. Nevertheless, the basal levels of ICaL were not significantly altered by chronic incubations with each of the following antioxidants: DMSO, glutathione, and N-acetylcysteine (a precursor of glutathione; see Supplementary material online, Figure S7). Therefore, the activity of pirfenidone as scavenger of ROS does not seem to be involved.
In addition to TGF-β1 and ROS, the following molecules were also ruled out as possible intermediaries, Ca2+ and TNF-α (Figure 4 and Supplementary material online, Figure S7). In particular, chronic treatments with Ca2+-chelant agents (BAPTA-AM and EGTA) were unable to mimic the potentiation of ICaL by pirfenidone, discarding that the drug acts by removing a potential Ca2+-dependent inactivation of CaV1.2 (see Supplementary material online, Figure S7). On the other hand, neither the basal levels of ICaL nor the response to pirfenidone were significantly altered by TNF-α (see Supplementary material online, Figure S7).
4. Discussion
The main conclusion of this study is that chronic pirfenidone treatment systematically and selectively increases the density of ICaL in both atrial and ventricular myocytes. To investigate the possibility that this effect could be due to higher expression of L-channels, we measured the amount of immobilization-resistance charge movement, which represents a reasonable estimation of the number of L-channels in the plasma membrane (assuming that the charge that moves per channel remains constant). As a result, we found that the average values of Qmax were unaltered. Accordingly, the levels of CaV1.2 mRNA and protein remained also unchanged. These data indicate that an enhanced function of CaV1.2 as the Ca2+ channel represents the underlying mechanism for the chronic potentiation of ICaL. Below, we discuss the potential pathophysiological relevance of our findings, the possible signalling pathways that may be involved, and certain limitations.
4.1 Signalling pathways
It is well known that molecules downstream of NO modulate not only L-channels but also other cardiac ion channels, in a species- and tissue-dependent manner (depending on their specific patterns of expression). One way requires that the NO bind to soluble guanylyl ciclase to activate it and increase levels of cyclic guanosine monophosphate (cGMP). Subsequently, cGMP can activate both cGMP-dependent protein kinase (PKG) and phosphodiesterase 2. The latter in turn inhibits the activity of L-channels, due to enhanced cAMP breakdown that ultimately results in reduced levels of PKA-dependent phosphorylation (for reviews, see refs22,23). As explained below, pirfenidone could have increased ICaL at least partially via chronic inhibition of this signalling pathway. For instance, inhibition of NOS expression or function is suggested not only by previous work4–6 but also from the time frame of their actions (i.e. reduces NOS expression and increases ICaL in ∼4 and ∼10 h, respectively). Additionally, the effect on ICaL was mimicked by an inhibitor of NOS (L-NAME). Furthermore, pirfenidone systematically produced an increase in both current density and voltage sensitivity, which is a typical consequence of CaV1.2 phosphorylation by PKA.24 Of course, given that the increase was not entirely suppressed by PKA inhibition, we anticipate that other mechanisms may be involved. For instance, pirfenidone may have prevented a potential direct inhibition of L-channels by NO and PKG, which act through S-nitrosylation25 and phosphorylation,26 respectively.
Conceivable, changes in subunit composition of the L-channel complex could also contribute. A higher ratio of CaV1.2 proteins interacting with β2a accessory subunits represents an intriguing mechanism. This is because β2a is almost unique in enhancing the amount of ICaL per channel;27 an effect due to a palmitoylation-dependent alosteric interaction,28 which enhances open probability and prevails over PKA-dependent modulation.29 Finally, another contributing factor could be palmitoylation of NOS, which has been shown to stimulate both NO production and targeting of the enzyme to plasmalemmal caveolae.30 Thus, it will be important for future studies to determine the possible impact of pirfenidone in expression and palmitoylation of cardiac NOS and β2a.
Pirfenidone exhibits anti-inflammatory, antioxidant, anti-fibrotic, and anti-proliferative properties, among others.1–3 With such a wide pharmacological profile, its high hydrophobicity (log P = 2.14) and low molecular weight (185 g/mol), it could be classified as a ‘promiscuous compound’, that is, compounds that genuinely bind to multiple defined molecular targets.31 This could explain why its mechanism of action is not fully understood.3 As a matter of fact, a systematic evaluation of cause–effect relationships is usually lacking in most of the studies, and thereby can only lead to speculation about potentially associated events. For instance, one may anticipate that the chronic or long-term actions could be the consequence of rapid antioxidant effects. However, in the literature, there is no direct evidence for this expectation. Thus, the great variety of chronic effects is most likely due to the binding of pirfenidone to another yet still unidentified molecular target(s) apart from ROS. In this regard, our finding that pirfenidone chronically modulates L-channels, independently of its activity as a scavenger of ROS, fits well with this interpretation. In general, long-term effects are explained by changes at the gene expression level. As a matter of fact, as we have been mentioning, pirfenidone alters the expression levels of a number of proteins, including NOS, TGF-β1, TNF-α, and other cytokines. Moreover, in hepatocytes, following a 2 h delay, pirfenidone inhibits transcriptional activation of the NOS gene promoter, and by 3–6 h of treatment, this results in both decreased expression of NOS protein and lower levels of NO production.4,5 Thus, it is tempting to speculate that the long time course for the onset of pirfenidone effect on ICaL could be due to a similar inhibition of NOS synthesis.
4.2 Pathophysiological relevance
The discovery of pirfenidone for potential use in cardiac therapy was based on its well-known anti-fibrotic activity. In particular, an Australian group found attenuation of cardiac fibrosis and stiffness in rat models of both diabetes and hypertension.9,10 Another Australian group, working with a mice model of Duchene muscular dystrophy reported that cardiac contractility is improved.13 More recently, a US group reported beneficial effects in canines with congestive heart failure (CHF),11 as well as in a rat model of myocardial infarct.12 In regard to the model of CHF, pirfenidone efficiently prevented the development of AF.11 This effect has been exclusively attributed to a significant reduction in atrial fibrosis.11 However, the following evidence suggests that it could be also partially explained by the present potentiation of L-channels. It is well known that the atrial electrical remodelling involves a significant reduction in ICaL (for a recent review, see Dobrev32). In fact, atrial ICaL is reduced in the same model33 as that in which pirfenidone prevented the development of AF,11 that is, in dogs with CHF induced by ventricular pacing. A lower density of ICaL, on the other hand, is considered an important substrate for the maintenance of AF.32 This is because it decreases both the APD and the effective refractory period (ERP), thereby allowing the accommodation of more APs per unit time.34,35 Conversely, as shown in Figure 6G, pirfenidone increases the APD, which in turn is expected to result in longer ERP and reduced AP frequency. Thus, it seems reasonable to speculate that the stimulation of L-channels may reinforce the ability of pirfenidone to prevent the development of AF. What is more, in human AF, a reduced ICaL has been associated with increased levels of S-nitrosylation of CaV1.2,36 and pirfenidone has the potential to both attenuate this abnormal increase in S-nytrosilation (by decreasing levels of NOS)4–6 and restore the normal density of atrial ICaL (this work).
In regard to the ventricle, an increase in ICaL could potentially result in the development of early after-depolarizations and arrhythmias.37 Interestingly, however, there is compelling evidence suggesting that pirfenidone is not adverse, but instead seems to be beneficial to ventricular function.9,10,12,13 For example, rats treated with pirfenidone are not only less prompt to develop ventricular tachycardia and infarction, but exhibit faster conducting velocities in their infarct border zone.12 Moreover, it has been reported that pirfenidone improves ventricular contractility in mice, even without a change in cardiac stiffness or fibrosis.13 Thus, there is room to speculate that the improvement of ventricular contractility represents a consequence of the higher activity of L-channels (via exacerbated Ca2+-induced Ca2+ release). Accordingly, pirfenidone drastically increases the force of contraction induced by a gradual exposure to elevated concentrations of extracellular Ca2+, but leaves intact the increase in force of contraction in response to noradrenaline.10 These observations are remarkably in line with our observation that the augmentation of ICaL produced by pirfenidone was blunted by two molecules downstream of noradrenaline receptors (i.e. cAMP and PKA; Figure 5B–D).
4.3 Potential limitations
Our results suggest that pirfenidone may activate the cAMP/PKA signalling. This conclusion is tentative and requires further investigation, as the activity and levels of these molecules were not measured in this study. It will be also important to investigate, in the near future, whether our present findings are also observed in human cardiac myocytes, since we did not perform experiments in cells from large animals. Likewise, it remains to be investigated whether an in vivo treatment with pirfenidone also results in a similar potentiation of ICaL. In regard to this issue, there is evidence suggesting that pirfenidone stimulates L-channels at clinically relevant concentrations: (i) within 3 h of a single oral administration of pirfenidone, the plasma concentration of the drug reaches levels higher than 40 µM, in both humans3 and rats;10 (ii) the corresponding steady-state levels decrease to ∼10 µM in rats that received oral pirfenidone for 14 days;10 and (iii) at these concentrations, that is, 40 and 10 µM, pirfenidone elicits 80 and 65%, respectively, of the maximum stimuli in L-channels (Figure 3C).
4.4 Conclusions
Our data suggest that pirfenidone stimulates the activity of a constant number of L-channels. This effect seems to involve long-term reduction of NO production, which apparently results in enhanced cAMP/PKA-dependent phosphorylation of CaV1.2. Whether these effects are also present in vivo, their precise pathophysiological consequences, and how they might contribute to justify the potential of pirfenidone as anti-arrhythmic still deserve to be systematically investigated.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by a Conacyt grant to G.A. (56733).
Acknowledgements
We thank Felipe Cruz Martinez and Mario Rodriguez Nieves for excellent technical assistance.
Conflict of interest: none declared.
References
Author notes
Present address. Carrera de Medicina, Facultad de Estudios Superiores de Iztacala, UNAM, Mexico.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}