





Abstract
Although a substantial role for 5′ adenosine monophosphate-activated protein kinase (AMPK) has been established in regulating cardiac metabolism, a less studied action of AMPK is its ability to prevent cardiac cell death. Using established AMPK activators like dexamethasone (DEX) or metformin (MET), the objective of the present study was to determine whether AMPK activation prevents tumour necrosis factor-alpha (TNF-α) induced apoptosis in adult rat ventricular cardiomyocytes.
Cardiomyocytes were incubated with DEX, MET, or TNF-α for varying durations (0–12 h). TNF-α-induced cell damage was evaluated by measuring caspase-3 activity and Hoechst staining. Protein and gene estimation techniques were employed to determine the mechanisms mediating the effects of AMPK activators on TNF-α-induced cardiomyocyte apoptosis. Incubation of myocytes with TNF-α for 8 h has increased caspase-3 activation and apoptotic cell death, an effect that was abrogated by DEX and MET. The beneficial effect of DEX and MET was associated with stimulation of AMPK, which led to a rapid and sustained increase in Bad phosphorylation. This event reduced the interaction between Bad and Bcl-xL, limiting cytochrome c release and caspase-3 activation. Addition of Compound C to inhibit AMPK reduced Bad phosphorylation and prevented the beneficial effects of AMPK against TNF-α-induced cytotoxicity.
Our data demonstrate that although DEX and MET are used as anti-inflammatory agents or insulin sensitizers, respectively, their common property to phosphorylate AMPK promotes cardiomyocyte cell survival through its regulation of Bad and the mitochondrial apoptotic mechanism.
1. Introduction
Tumour necrosis factor-alpha (TNF-α), expressed as a 26 kDa membrane bound protein, is cleaved by TNF-α converting enzyme to produce a soluble 17 kDa biologically active protein.1,2 Primarily produced by macrophages,3,4 TNF-α is also synthesized by a variety of other cell types including lymphoid,5 endothelial,6 mast cells,7 adipose8 and neuronal tissues,9 fibroblasts,10 and cardiac myocytes.11 The effects of TNF-α are mediated by its binding to two distinct cellular receptors, TNFR1 and TNFR2. Although stimulation of cells with TNF-α regulates numerous cellular and biological processes such as immune function, cell proliferation, differentiation, and energy metabolism,12 binding of excess TNF-α to TNFR1 activates the programmed cell death (apoptosis) pathway. This includes formation of the death-inducing signalling complex, activation of the caspase cascade,13 impairment of mitochondrial integrity, cytochrome C release,14 and eventually apoptosis in a multitude of cells including fibroblasts, vascular smooth muscle, endothelial cells, and cardiomyocytes.
Elevated plasma or endogenous cardiac TNF-α levels have been reported to cause myocyte apoptosis,15 leading to cardiomyocyte loss with associated cardiovascular diseases. For example, transgenic mice with cardiac-specific overexpression of TNF-α demonstrate increased apoptosis and develop cardiac hypertrophy and dilated cardiomyopathy.16,17 Stress can cause cardiomyocytes and resident macrophages to produce TNF-α that binds to myocardial TNFR1 and induces apoptosis. Augmented plasma TNF-α levels and increased expression of TNFR1 in the arterial wall have been found to promote atherosclerosis.18 This cytokine has also been suggested to contribute towards cardiomyocyte cell death and cardiac dysfunction in clinical conditions like sepsis,19 chronic heart failure, and ischaemia–reperfusion injury,20 and cardiac allograft rejection.21 Till date, a variety of apoptotic mediators have been proposed to facilitate TNF-α induced cardiomyocyte apoptosis, which include ceramides,22 nitric oxide,23 and mitogen-activated protein kinases.24 Regulation of myocardial apoptosis is also achieved by the Bcl-2 family of proteins;25 Bad, which initiates apoptosis-induced signalling, and Bcl-xL that blocks activation of effector caspases and induction of apoptosis.
The fuel gauge AMP-activated protein kinase (AMPK) facilitates ATP production to meet energy demands during metabolic stress.26 Allosteric binding of AMP or phosphorylation by upstream kinases (tumour suppressor kinase—LKB1; Ca2+ calmodulin-dependent protein kinase kinases—CaMKK) activates AMPK.27 Once activated, AMPK facilitates substrate delivery and subsequent oxidation to generate ATP. Although a substantial role for AMPK has been established in regulating cardiac metabolism, a less studied action of AMPK is its ability to prevent cell death. In non-cardiac cells like endothelial cells or astrocytes, activation of AMPK has been shown to protect against hyperglycaemia or high fat induced apoptotic cell death.28,29 In H9c2 rat cardiac muscle cell lines or adult cardiomyocytes, AMPK activation protects against reactive oxygen species or high fat induced cell death.30 Using established AMPK activators like dexamethasone (DEX) or metformin (MET), the objective of the present study was to determine whether AMPK activation prevents TNF-α-induced apoptosis in adult rat ventricular cardiomyocytes. Our data demonstrate that by phosphorylating Bad and maintaining the anti-apoptotic property of Bcl-xL, AMPK confers protection against TNF-α-induced apoptosis.
2. Methods
2.1 Experimental animals
The investigation conforms with the guide for the care and use of laboratory animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and the University of British Columbia. Adult male Wistar rats (260–300 g) were obtained from the UBC Animal Care Unit and given free access to a standard laboratory diet (PMI Feeds, Richmond, VA, USA) and water.
2.2 Isolation of cardiomyocytes
Ventricular myocytes were prepared by a previously described procedure.31 Where indicated, DEX (100 nM), MET (2 mM) (we have previously reported that concentrations of DEX and MET were optimal to activate AMPK),32,33 or TNF-α (0–100 ng/mL) were added to the culture medium and myocytes kept for varying durations (0–12 h). At the indicated times, cells were used for evaluation of intracellular calcium and apoptotic cell death. In separate experiments, cells were scraped and washed twice with 0.5 mL PBS. Samples were lysed in ice-cold buffer and myocyte cell lysates were used for western blot, immunoprecipitation, and gene estimations. Where indicated, STO609 (2 µM), RU486 (10 µM), or Compound C (40 µM) were used to inhibit CAMKKβ, glucocorticoid receptor, or AMPK, respectively. Compound C is a cell-permeable, selective, reversible, and competitive inhibitor of AMPK.
2.3 Estimation of cardiomyocyte intracellular free Ca2+
Ca2+ was measured using the Fura 2-AM method as described previously.34
2.4 Measurement of apoptosis
To examine TNF-α-induced apoptosis and the influence of DEX and MET on this process, cells were examined for (a) morphological evidence of apoptosis using the fluorescent DNA-binding dye, Hoechst 33342 (Sigma) or (b) changes in caspase-3 (enzyme that plays a key role in apoptosis) activity using an assay kit or western blot. Caspase-3 activity is presented as activity per milligram of protein for each sample. Caspase-3 activation was also confirmed by measuring cleaved caspase-3 using western blot.
2.5 Separation of mitochondrial and cytosolic fractions
Cardiomyocytes were collected in ice-cold PBS, spun down, resuspended in cytosol extraction buffer mix, vortexed and incubated on ice for 10 min. Samples were centrifuged at 2600 rpm (700 g) for 10 min and the supernatant transferred and spun at 9600 rpm (10 000 g) for 30 min to precipitate mitochondria. Supernatant was removed as the cytosolic fraction and the pellet were resuspended in mitochondrial extraction buffer mix as the mitochondrial fraction.
2.6 Western blot
Western blot was carried out as described previously.35
2.7 Immunoprecipitation
Cell lysates were immunoprecipitated using a Bad monoclonal antibody rotating overnight at 4°C. The immunocomplex and the supernatant were resuspended in Laemmli buffer and heated for 5 min at 95°C. The immunocomplex was separated into two equal portions, each of which was immunoblotted with anti-Bcl-xL and anti-Bad using western blot.
2.8 Cardiomyocyte AMPK gene expression
AMPK gene expression was measured in cardiac cells using RT–PCR.36
2.9 Materials
Total AMPK-α, phospho-AMPK-α (Thr172), phospho ACC, Bcl-xL, GAPDH, prohibitin (PHB1), cleaved caspase-3 (Asp175), and caspase-3 antibodies were obtained from Cell Signaling (Danvers, MA, USA). Total Bad, phospho-Bad (Ser112), Total CaMKI, phospho-CaMKI (Thr177), and cytochrome C antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). EnzChek Caspase-3 Assay Kit was obtained from Molecular Probes (Eugene, OR, USA). The CAMKKβ inhibitor STO609 was obtained from Calbiochem (La Jolla, CA, USA). All other chemicals were obtained from Sigma Chemical.
2.10 Statistical analysis
Values are means ± SE. Wherever appropriate, one-way ANOVA followed by Tukey's or Bonferroni tests to determine differences between group mean values. The level of statistical significance was set at P < 0.05.
An expanded version of the materials and methods has been described in the online Supplementary material.
3. Results
3.1 Influence of DEX on total and phosphorylated AMPK in isolated cardiac myocytes
Previous studies from our laboratory have reported an increased phosphorylation and activation of AMPK in hearts from 4 h DEX-treated animals.37 To assess direct activation of AMPK, control cardiomyocytes were incubated with DEX for varying times. In the absence of DEX, no change in AMPK phosphorylation was observed over time (data not shown). DEX induced a time-dependent increase in AMPK phosphorylation, an effect that peaked at 2 h and was maintained up to 8 h (Figure 1A). Once activated, AMPK phosphorylates and inactivates ACC. Similar to AMPK, ACC phosphorylation increased temporally in DEX-treated cardiomyocytes (Figure 1A, immunoblot). The increase in AMPK phosphorylation (up to 4 h) following DEX could not be explained by any changes in AMPK total protein or gene expression. An augmentation in total AMPK protein and gene expression became apparent only 8 h after DEX incubation (Figure 1B and C).
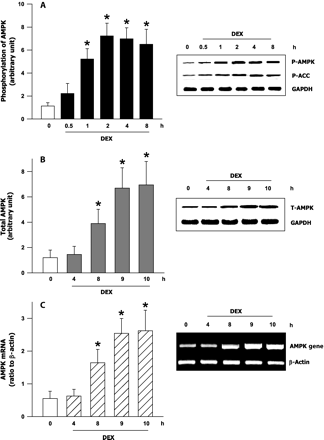
Time-dependant changes in AMPK in control and DEX-treated cardiomyocytes. Cardiomyocytes were plated on laminin-coated culture plates. Cells were maintained using Medium 199 and incubated at 37°C under an atmosphere of 95% O2–5% CO2 for 16 h. Subsequently, DEX (100 nM) was added to the culture medium. At the indicated times, cells were scraped and protein extracted to determine phosphorylated AMPK (A) and ACC (A, immunoblot), and total AMPK (B) using western blotting. AMPK gene expression was evaluated using RT–PCR (C). Results are mean ± SE of five myocyte preparations in each group. Asterisks denote significantly different results from untreated control (0 min), P < 0.05. DEX, dexamethasone; P, phosphorylated.
3.2 Changes in cardiomyocyte intracellular calcium and CAMKI phosphorylation following DEX
Elevation in cytosolic calcium increases the phosphorylation of CaMKI (Thr177), an upstream regulator of AMPK.38,39 To determine the mechanism by which DEX induces the early activation of AMPK, we measured intracellular calcium and CaMKI phosphorylation. Cells treated with DEX displayed a robust increase in intracellular calcium (Figure 2A). Peak responses were recorded within 5 min, and were maintained up to 25 min after DEX. Although the calcium response declined over time, it was maintained above baseline up to 1 h after DEX (Figure 2A). CaMKI (Thr177) phosphorylation followed the rise in intracellular calcium observed with DEX; an increased phosphorylation was observed within 15 min of DEX incubation (Figure 2B). Interestingly, CAMKI phosphorylation peaked at 60 min followed by a return to near control levels after 8 h of DEX (Figure 2B). No change in total CaMKI protein was observed with DEX treatment (Figure 2B, immunoblot).
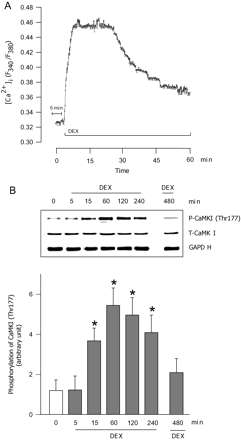
Effects of DEX on cardiomyocyte intracellular Ca2+ and CAMKI phosphorylation. In cardiomyocytes exposed to DEX (100 nM), changes in cytosolic Ca2+ over time were measured using Fura 2 (A). Horizontal bars indicate an initial baseline period of 5 min followed by DEX treatment for 1 h. The panel indicates an average response of 15–25 cardiac cells of 4 separate myocyte preparations. To evaluate the effects of DEX on CAMKI phosphorylation, laminin-plated cardiomyocytes were exposed to DEX (100 nM). At the indicated times (0–480 min), protein was extracted to determine phosphorylation of CaMKI (Thr177) (B) and total CaMKI (B, immunoblot). Results are means ± SE of five myocyte preparations in each group. Asterisks denote significantly different results from untreated control (0 min), P < 0.05. DEX, dexamethasone.
3.3 Dual regulation of AMPK phosphorylation by DEX
To validate the impact of CAMKI on AMPK phosphorylation, cardiomyocytes were treated with DEX in the presence or absence of STO609, a specific inhibitor of CaMKKß, an upstream regulator of CaMKI. STO609 prevented the DEX induced phosphorylation of CAMKI (Figure 3A) and AMPK (Figure 3B) at 2 h, an effect that was absent when the incubation with DEX was extended to 8 h (Figure 3C). By itself, STO609 had no effect on CaMKI or AMPK phosphorylation (data not shown). Interestingly, the glucocorticoid receptor antagonist RU486 did not influence the early (2 h) impact of DEX on CaMKI (Figure 3A), or AMPK phosphorylation (Figure 3B), suggesting that at this time, the effects of DEX are likely mediated by calcium. However, at 8 h, RU486 abrogated the DEX-induced increase in AMPK phosphorylation (Figure 3C) and total AMPK and gene expression (Figure 3C, immunoblots).
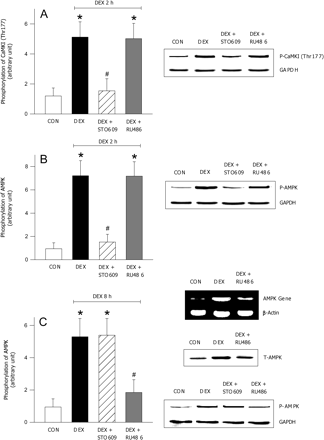
Inhibition of AMPK activation induced by DEX. Laminin-plated cardiomyocytes were incubated with 100 nM DEX for 2 h (A and B) or 8 h (C). Where indicated, cells were pre-incubated for 45 min with STO609 (2 µM) or RU486 (10 µM) prior to addition of DEX. Total protein and RNA were extracted to determine phosphorylation of CAMKI (Thr177) (A) and AMPK (B and C), total AMPK (C, immunoblot) and AMPK gene expression (C, immunoblot). Results are means ± SE of five myocyte preparations in each group. Asterisks denote significantly different results from untreated control (CON), hash symbol indicates significantly different results from DEX treated, P < 0.05. DEX, dexamethasone; P, phosphorylated.
3.4 TNF-α-mediated effects on cardiomyocyte cell death
An initial experiment was performed to determine the concentration and time required for TNF-α to induce optimal cardiomyocyte cell death. Cardiomyocytes were exposed to various concentrations of TNF-α (0–100 ng/mL) and caspase-3 activity determined. As concentrations of TNF-α between 10 and 100 ng/mL produced comparable levels of caspase-3 activation (Figure 4A), a dose of 10 ng/mL was used for all subsequent experiments. Using this concentration of TNF-α, a time-dependant assay revealed similar levels of caspase-3 activation (Figure 4B) and apoptotic cell death (Figure 4C and see Supplementary material online, Figure S1) after 8 and 12 h. Thus, 8 h was chosen as the optimum period for all subsequent experiments utilizing TNF-α.
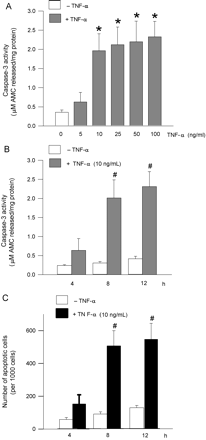
TNF-α induced cardiomyocyte apoptosis. (A) Ventricular cardiomyocytes were exposed to various concentrations of TNF-α (0–100 ng/mL) for 8 h and caspase-3 activity measured. To evaluate time-dependant effects, cardiomyocytes were incubated with 10 ng/mL TNF-α for 4, 8, and 12 h and caspase-3 activity (B) and evidence of apoptosis using the fluorescent DNA-binding dye Hoechst 33342 (C) were determined. Results are means ± SE of 4–5 myocyte preparations in each group. Asterisks denote significantly different results from untreated control (0 min), hash symbol indicates significantly different results from 4 h TNF-α treated, P < 0.05.
3.5 AMPK regulation of TNF-α-mediated caspase-3 activation and cell death
Metformin is an established activator of AMPK in cardiomyocytes.33 Treatment of control myocytes with DEX or MET for 8 h resulted in an increase in AMPK phosphorylation, with DEX displaying an approximately two-fold greater AMPK activation when compared with MET (Figure 5A). Interestingly, the DEX-induced changes in AMPK phosphorylation were accompanied by an increase in total AMPK protein, an effect that was absent with MET (Figure 5A, immunoblot). Although TNF-α by itself marginally suppressed AMPK phosphorylation, both DEX and MET were still able to activate AMPK in the presence of this cytokine (Figure 5A). TNF-α had no influence on total AMPK protein, either by itself or in the presence of DEX (Figure 5A, immunoblot). Both DEX and MET induced a robust reduction in the TNF-α-induced activation of caspase-3 (Figure 5B and C). Figure 5D describes the protective effects of these AMPK activators on TNF-α-induced cardiomyocyte cell death. In the control group, less than 60 of 1000 cells were scored as apoptotic. TNF-α significantly increased apoptosis (623 of 1000 cells). Introducing DEX and MET into the medium significantly lowered the number of apoptotic cells (DEX, 182 of 1000; MET, 196 of 1000 cells). Counting the total number of surviving cells revealed that with TNF-α, a survival rate of 39% was observed. Addition of DEX and MET increased the survival of cardiomyocytes to 83 and 84%, respectively. These data suggest that under our experimental conditions, the TNF-α-induced cell death is predominantly apoptotic in nature.
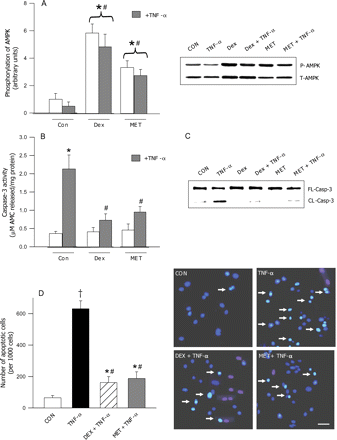
Effects of AMPK activation on TNF-α-induced apoptosis. Cardiomyocytes were incubated with DEX (100 nM) or metformin (2 mmol/L) in the presence or absence of 10 ng/mL TNF-α. Following 8 h of incubation, cells were scraped and protein extracted to determine phosphorylated AMPK (A) and total AMPK (A, immunoblot). Caspase-3 activity (B) and cleavage (C) were measured using a fluorometric assay kit or western blotting. Cells were also examined for morphological evidence of apoptosis using the fluorescent DNA-binding dye Hoechst 33342 (D). Results are means ± SE of five myocyte preparations in each group. Bar = 25 µm. Asterisks denote significantly different results from control (CON), hash indicates significantly different results from TNF-α treated (without DEX or MET), dagger indicates significantly different results from all other groups, P < 0.05. DEX, dexamethasone; MET, metformin; P, phosphorylated; T, total; FL, full length; CL, cleaved.
In vivo, DEX after 4 h increased AMPK phosphorylation, an effect that was maintained until 8 h (see Supplementary material online, Figure S2A). This change in AMPK phosphorylation paralleled a rise in total AMPK protein and gene expression (see Supplementary material online, Figure S2A, insets). Interestingly, AMPK remained activated in myocytes from DEX-treated animals, an effect that remained unchanged on addition of TNF-α for 8 h (see Supplementary material online, Figure S2B). More importantly, the protective effect of DEX against TNF-α-induced caspase activation was maintained in these myocytes even in the direct absence of DEX in the culture media (see Supplementary material online, Figure S2C).
3.6 DEX-induced effects on Bad phosphorylation and cytochrome C release
The pro-apoptotic protein Bad plays a significant role in activating the caspase cascade of cell death. To determine whether DEX inhibits TNF-α-induced apoptosis by phosphorylating and inactivating Bad, we examined Bad phosphorylation in control myocytes treated with DEX. Phospho Bad (Ser112) levels were significantly higher by 30 min, reached near maximal values by 2 h, and remained activated up to 8 h after DEX treatment (Figure 6A). No change in total Bad protein was observed over time (Figure 6A, immunoblot). Bad phosphorylation reduces its association with Bcl-xL, thus maintaining the anti-apoptotic function of this protein. In the presence of TNF-α, Bad phosphorylation was reduced (Figure 6B, immunoblot), with a corresponding increase in its interaction with Bcl-xL (Figure 6B). Addition of DEX to the incubation medium resulted in a dramatic loss of association between Bad and Bcl-xL (Figure 6B), and was consistent with a phosphorylation-induced dissociation (Figure 6B, immunoblot). Release of mitochondrial cytochrome C to the cytosol is a critical step in the mechanism of TNF-α-induced apoptotic cell death. Figure 6C shows a dramatic increase in cytosolic cytochrome C levels in TNF-α-treated myocytes, an effect that was prevented by addition of DEX.
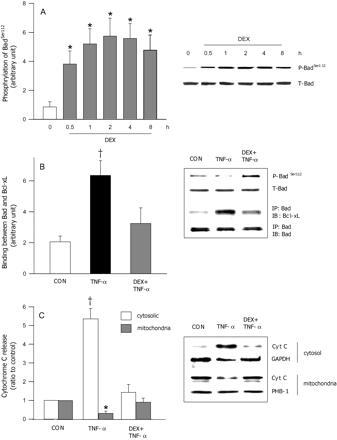
DEX-induced regulation of Bad phosphorylation and its association with BCL-xL. In cardiomyocytes treated with DEX (100 nM), time-dependant changes in Bad phosphorylation (Ser112) (A) and total Bad (A, immunoblot) were determined using western blotting. To determine the effects of DEX on TNF-α-induced changes in total and phosphorylated Bad (Ser112), ventricular cardiomyocytes were incubated with 10 ng/mL TNF-α for 8 h, in the presence or absence of DEX (100 nM) (B, immunoblot). To examine the association between Bad and Bcl-xL in these cells, Bad was immunoprecipitated (IP) using a Bad antibody and immunoblotted (IB) with anti-Bcl-xL (B). Cytochrome c release was also determined in these cells by preparing cytosolic and mitochondrial extracts as described in Methods section. The samples were then subjected to western blot analysis for cytochrome c (C). Prohibitin-1 (PHB 1) was immunoblotted as a mitochondrial marker (C, immunoblot). Results are means ± SE of four myocyte preparations in each group. Asterisks denote significantly different results from control (CON), dagger indicates significantly different results from all other groups, P < 0.05. DEX, dexamethasone; P, phosphorylated; T, total.
3.7 Consequence of AMPK inhibition on Bad phosphorylation and TNF-α-induced caspase-3 activation
To confirm whether DEX-induced AMPK activation influences Bad phosphorylation, we used Compound C to inhibit AMPK phosphorylation. Pre-incubation of cardiomyocytes with Compound C abolished the DEX-induced activation of AMPK (Figure 7A). Compound C had no effect on the DEX-induced augmentation of total AMPK protein (Figure 7A, immunoblot). Interestingly, this decrease in AMPK phosphorylation led to a substantial reduction in Bad (Ser112) phosphorylation (Figure 7B). To evaluate the effects of AMPK inhibition on TNF-α-mediated caspase-3 activation, cells were treated with TNF-α, DEX, and Compound C. Addition of Compound C to cells treated with DEX+TNF-α reduced phosphorylation of AMPK (Figure 7C) and Bad (Figure 7C, immunoblot), increased the association between Bad and Bcl-xL (Figure 7D), and provoked caspase-3 activation (Figure 7E).
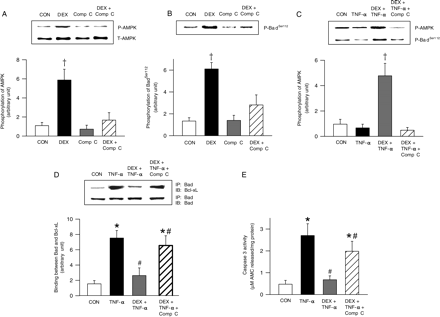
Consequence of AMPK inhibition on TNF-α-induced Bad phosphorylation and caspase-3 activity. Cardiomyocytes were pre-incubated with Compound C (40 µM) for 45 min, prior to DEX (100 nM) treatment for 8 h. Cells were scraped and protein extracted to determine total (A, immunoblot) and phosphorylated (A) AMPK, and phosphorylated Bad (Ser112) (B). To evaluate the effects of AMPK inhibition on TNF-α-mediated effects on cardiomyocytes, cells were exposed to TNF-α (10 ng/mL) or DEX (100 nM), or their combination for 8 h. Where indicated, one group was subjected to a 45 min pre-exposure to Compound C followed by incubation with DEX and TNF-α for 8 h. Cells were scraped and protein extracted to determine the phosphorylation of AMPK (C) and Bad (Ser112) (C, immunoblot). Cell lysates from the above groups were also used to evaluate the interaction between Bad and Bcl-xL using immunoprecipitation (D). Caspase-3 activity was measured using a fluorometric assay kit (E). Results are means ± SE of four myocyte preparations in each group. Asterisks denote significantly different results from control (CON), hash indicates significantly different results from TNF-α, double dagger symbols denote significantly different results from DEX + TNF-α treates, dagger symbols denote significantly different results from all other groups, P < 0.05. DEX, dexamethasone; P, phosphorylated; T, total; IP, immunoprecipitated; IB, immunoblot.
3.8 Cardioprotective effects of MET against TNF-α-induced caspase-3 activation
Metformin time dependently stimulated AMPK phosphorylation without changing total AMPK protein (see Supplementary material online, Figure S3A). In the presence of TNF-α, MET like DEX: (a) increased Bad phosphorylation (see Supplementary material online, Figure S3B, inset), (b) reduced the association between Bad and Bcl-xL (see Supplementary material online, Figure S3B), (c) decreased cytochrome c release (see Supplementary material online, Figure S3C), and (d) significantly lowered caspase-3 activation (see Supplementary material online, Figure S3D). Pre-incubation of cardiomyocytes with Compound C prevented all of these effects (see Supplementary material online, Figure S4A–E). Collectively, our results imply that once AMPK is activated by DEX or MET, similar mechanisms are turned on to protect against TNF-α-induced cell death.
4. Discussion
TNF-α has been reported to induce apoptosis in numerous cell types through its activation of a variety of signalling pathways. In cardiac myocytes, these include increased expression of iNOS,40 production of ceramides,22 and activation of the mitogen-activated protein kinase (MAPK) pathway.24 Limited information is available that documents signalling mechanisms that protect myocytes against TNF-α-induced apoptosis. Our data suggest that activation of AMPK using DEX or MET protects adult cardiomyocytes against TNF-α-induced apoptosis by phosphorylating Bad, thereby preventing cytochrome c release and attenuating caspase-3 activation.
In cardiac cells, AMPK is the switch that regulates cellular energy during metabolic stress.26 Activation of AMPK can occur as a consequence of a rise in the AMP/ATP ratio (when ATP is depleted) or through its regulation by upstream kinases like LKB1 and CaMKKβ.27 Glucocorticoids such as DEX, which are potent anti-inflammatory drugs used for the treatment of a number of human diseases, are believed to act via both non-genomic (rapid) and genomic pathways.41 In the present study, DEX treatment of cardiomyocytes led to an early (within 1 h) increase in AMPK phosphorylation that was sustained up to 8 h after DEX. Although this early increase in AMPK phosphorylation may well be a product of the rapid action of glucocorticoids to compromise ATP production, that highlights its actions as an immunosuppressant to reduce inflammatory processes,42 this is unlikely to occur in the heart that is fundamentally dependant on this nucleotide for its normal functioning. In the absence of this mechanism, a possible candidate to explain the early activation of AMPK is calcium. Elevation of intracellular calcium activates CaMKKβ, which directly phosphorylates CaMKI (Thr177), an upstream regulator of AMPK.38,39,43 Glucocorticoids transiently increases Ca2+ via non-genomic pathway in many cell types including brain synaptosomes44 and cultured hippocampal44 cells. In the heart, DEX treatment improves the reduced myocardial function and metabolism in acute myocardial infarction by its influence on intracellular Ca2+ levels.45 In rat neonatal cardiomyocytes, DEX augments intracellular calcium levels by increasing the activity of L-type calcium channels.46 Given this established role of DEX in influencing cellular calcium levels, we measured and document a rapid and transient increase in intracellular calcium followed by a rise in CaMKI on in vitro exposure of cardiomyocytes to this glucocorticoid. As STO-609 (a selective cell-permeable inhibitor of CaMKK), and not RU486 (a glucocorticoid receptor antagonist), inhibited the activities of CaMKI and AMPK, our data suggest that early activation of AMPK by DEX is independent of receptor stimulation and likely mediated through a calcium/CaMKI pathway. Interestingly, AMPK activation by DEX was maintained up to 8 h, even though intracellular calcium declined, and CaMKI phosphorylation returned to near normal levels. As glucocorticoids act predominantly through genomic mechanisms that include binding to a cytosolic receptor, relocation into the nucleus, and an increase or decrease in gene expression,47 we measured AMPK protein and gene expression, and report an increase only after 8 h of DEX. Given that RU486 and not STO-609 prevented the DEX-induced increases in AMPK protein and gene expression and phosphorylation at this time, our data imply that activation of AMPK at 8 h is receptor mediated and genomic in nature.
Cardiac failure induced by TNF-α is either a consequence of direct reduction in contractility48 or stimulation of myocyte apoptosis.49 In this study, appreciable activation of caspase-3 and evidence of apoptosis in rat cardiomyocytes occurred at a concentration of 10 ng/mL of TNF-α and within 8 h of incubation. Using different cell types, other studies have also reported similar concentration and time-dependant effects of TNF-α on this mode of cell death.50 To evaluate the influence of AMPK activation on protecting cardiomyocytes against TNF-α-mediated apoptosis, we used DEX or MET. Interestingly, when compared with MET, AMPK phosphorylation in myocytes treated with DEX was approximately two-fold higher, and probably was a reflection of both the non-genomic and genomic actions of DEX. Activation of AMPK with DEX or MET was associated with a robust reduction in TNF-α-induced caspase-3 activation and apoptotic cell death. As these effects were apparent even with a disparate activation of AMPK, our data suggest that a threshold for AMPK activation likely exists to suppress the action of TNF-α on apoptosis. In support of these findings, other studies have also documented beneficial effects of AMPK activation against other initiators of cell death. For example, in H9c2 cardiac cells, AMPK activation has been shown to protect against reactive oxygen species-mediated cell death.30 In osteoblasts, AICAR by activating AMPK has been shown to inhibit palmitate-induced apoptosis.51 In adult cardiomyocytes, we have reported that activation of AMPK by MET protects against palmitate-induced cell death.33 Other studies have also reported that MET confers cardioprotection against myocardial infraction and heart failure by activating AMPK.52,53 Overall, our data suggest that although these drugs are used as anti-inflammatory agents (DEX) or insulin sensitizers (MET), their common property to phosphorylate AMPK confers protection against cytokine-induced cardiac cell death.
The function of Bad, a pro-apoptotic protein, is tightly regulated by phosphorylation. Bad phosphorylation facilitates its association with 14:3:3 proteins and prevents its translocation to the mitochondria. This event limits its interaction with the anti-apoptotic protein Bcl-xL, allowing the latter protein to promote cell survival.24 Maintenance of Bad phosphorylation has been reported to prevent apoptosis of rat hepatic sinusoidal endothelial cells.54 In failing hearts, a significant decrease in phosphorylated Bad (Ser112) has been reported to initiate apoptosis.55 TNF-α is known to decrease Bad phosphorylation, augment its association with Bcl-xL, and promote cytochrome c release and caspase-3 activation,56 effects that were duplicated in the present study. DEX treatment of cardiomyocytes led to a rapid and sustained increase in the phosphorylation of Bad, an effect that was comparable to its influence on AMPK phosphorylation. As compound C prevented the DEX-induced activation of AMPK and phosphorylation of Bad, our data suggest that AMPK is an upstream regulator of Bad. More importantly, the effect of DEX on Bad phosphorylation reduced the TNF-α-mediated association b6etween Bad and Bcl-xL, and prevented the release of cytochrome c (Figures 6 and 8). As compound C abrogated all of these effects, our data suggest that in addition to its previously reported modulation of cardiac metabolism, AMPK can promote cardiomyocyte cell survival through its regulation of Bad and the mitochondrial apoptotic mechanism. It should be noted that activation of AMPK by the concentration of DEX (100 nM) used in the present study prevented only cell death induced by TNF-α, and not other death-inducing stimuli like IL-1β or excess epinephrine (data not shown). At present, the mechanisms behind this selective protection of DEX against TNF-α-induced apoptosis are unclear but could be related to dissimilar mechanisms that initiate cell death. IL-1β is suggested to trigger apoptosis through stimulation of nitric oxide and peroxynitrite production,57 whereas excess epinephrine-induced apoptosis is suggested to be a result of a number of mechanisms that involve both β-adrenergic- (reactive oxygen species and calcium overload) and non-β-adrenergic- (accumulation of pro-apoptotic catecholamine metabolites) specific mechanisms.58 Additionally, this lack of protection could also be related to the concentration of DEX used when utilizing IL-1β or excess epinephrine to initiate apoptosis, with higher concentrations likely offering some cardioprotection. Whether MET can also prevent cell death induced by IL-1β or excess catecholamine is unknown, and warrants further investigation.
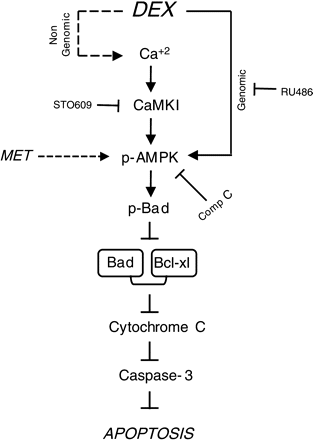
Graphic representation of the proposed mechanism mediating the protective effect of AMPK activation on TNF-α-induced apoptosis in adult rat ventricular cardiomyocytes. DEX (or MET), by activating AMPK, either through a Ca2+/CaMKI/AMPK pathway and receptor-mediated increase in gene expression, phosphorylates and inactivates Bad, prevents cytochrome c release, and inhibits activation of the caspase cascade and apoptotic cell death.
In summary, irrespective of the mechanism by which AMPK is phosphorylated, its activation is crucial for imparting protection to the cardiomyocyte against TNF-α-induced cytotoxicity. As the production of TNF-α is widely reported to increase during obesity, diabetes,59 and ischaemia reperfusion,60 activation of AMPK in these conditions may protect against this specific cytokine-induced cardiac injury.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
The studies described in this paper were supported by an operating grant from the Heart and Stroke Foundation of BC and Yukon.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}