





Abstract
Mitochondrial dysfunction is a major factor in heart failure (HF). A pronounced variability of mitochondrial electron transport chain (ETC) defects is reported to occur in severe acquired cardiomyopathies without a consistent trend for depressed activity or expression. The aim of this study was to define the defect in the integrative function of cardiac mitochondria in coronary microembolization-induced HF.
Studies were performed in the canine coronary microembolization-induced HF model of moderate severity. Oxidative phosphorylation was assessed as the integrative function of mitochondria, using a comprehensive variety of substrates in order to investigate mitochondrial membrane transport, dehydrogenase activity and electron-transport coupled to ATP synthesis. The supramolecular organization of the mitochondrial ETC also was investigated by native gel electrophoresis. We found a dramatic decrease in ADP-stimulated respiration that was not relieved by an uncoupler. Moreover, the ADP/O ratio was normal, indicating no defect in the phosphorylation apparatus. The data point to a defect in oxidative phosphorylation within the ETC. However, the individual activities of ETC complexes were normal. The amount of the supercomplex consisting of complex I/complex III dimer/complex IV, the major form of respirasome considered essential for oxidative phosphorylation, was decreased.
We propose that the mitochondrial defect lies in the supermolecular assembly rather than in the individual components of the ETC.
1. Introduction
The heart has a continuous requirement for energy in the form of ATP. Mitochondria are the primary ATP provider via the conversion of food fuels to usable energy. Compelling evidence for a link between the decline in mitochondrial energy transduction and mechanical dysfunction in heart failure (HF) is found in animal models and in patients with genetic mitochondrial diseases1 or acquired cardiomyopathies.2–4
Although HF of different aetiology has common cardiac mechanical dysfunctions, the investigation of mitochondrial function in humans and animal models of HF shows a variety of mitochondrial alterations5 that point to defects at specific sites in mitochondrial energy transfer pathways, i.e. the electron transport chain (ETC)6 or the phosphorylation apparatus.7 However, no comprehensive examination of cardiac mitochondrial function has been reported in HF.
Studies performed in severe HF show mitochondrial dysfunction, whereas data generated in models with moderate severity HF are controversial.5 These conflicting data may be explained by differences in the approach used by different laboratories to assess mitochondrial function. For example, the oxygen consumption of heart homogenate was preserved until late stages8 in pressure and volume overload-induced HF. In contrast, isolated mitochondria from both pressure-overload9 and genetic cardiomyopathic hamster hearts10 exhibited a progressive decline in oxidative phosphorylation with the severity of HF. Similarly, mitochondrial respiratory activity was decreased in cardiomyocytes isolated from humans and dogs with chronic HF,2,11,12 even though the mitochondrial volume density and activity of citrate synthase were normal.13,14 Defects in mitochondrial function may not be detected in the early stages of HF when measured in muscle homogenates or fibres due to possible alterations in mitochondrial density15 and heterogeneity.16 Moreover, heart mitochondria exist as two functionally and biochemically distinct populations: subsarcolemmal mitochondria (SSM), which are situated beneath the plasma membrane, and interfibrillar mitochondria (IFM), which are located among the myofibrils.17 Most studies on mitochondrial respiration in HF were performed on the combined populations of heart mitochondria released by polytron homogenization and protease treatment.9,16 The two heart mitochondrial populations may be affected differently in HF because they are biochemically distinct17 and they react differently in pathological states such as aging18,19 or genetic cardiomyopathy.10
The goal of the current study was to evaluate the integrative function of SSM and IFM in moderately severe HF. Studies were performed in the well-established canine coronary microembolism-induced HF model.12,14,20 To this end, oxidative phosphorylation, as an integrative function of mitochondria, was assessed with a comprehensive number of substrates in order to investigate membrane transport, dehydrogenase activities, and the ETC coupled to ATP synthesis. We found a dramatic decrease in ADP-stimulated respiration that was not relieved by an uncoupler. Moreover, the ADP/O ratio was normal, indicating no effect on the phosphorylation apparatus. The data point to a defect in oxidative phosphorylation within the ETC. However, the individual activity of ETC enzymes was normal. The amount of the supercomplex consisting of complex I/complex III dimer/complex IV, the major form of respirasome considered essential for oxidative phosphorylation, was decreased. In contrast, the amount of free complexes I and III were increased in SSM. In summary, there is a dramatic decrease in oxidative phosphorylation of heart mitochondria isolated from moderately severe HF, which is associated with a decrease in the content of ETC respirasome.
2. Methods
Please see the online supplement for additional experimental details for several of the methods mentioned here.
2.1 Materials, reagents
Unless specified, all reagents were purchased from Sigma-Aldrich (St Louis, MO, USA) and were of the highest purity grade.
2.2 Animal model
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All experimental protocols were approved by the Henry Ford Hospital and Case Western Reserve University Institutional Animal Care and Use Committees. The intracoronary microembolization-induced model was used to induce HF in dogs, as described.20,21 A single intracoronary artery injection of polystyrene latex microspheres (70–102 µm in diameter) was administered 1–2 weeks apart for four to six times by alternative catheterization of either left anterior descending or circumflex arteries causing left ventricle (LV) microinfarctions. Embolizations were discontinued when LV ejection fractions were <35%. Dogs were maintained for a period of 1 week to ensure that infarctions produced by the last procedure were completely healed. Ventriculograms were performed to measure LV function prior the first embolization and 1 week after the last microembolization. Two weeks after the last ventriculogram, dogs were anesthetized (sodium thiopental 25 mg/kg iv followed by isoflurane 0.75–1.5%), the heart exposed, and tissue from the free wall of the LV collected. The control group consisted of five age-matched control dogs that did not undergo either the cardiac catheterization or the multiple sequential intracoronary microembolization procedure, and were studied at the same time and were monitored clinically and biochemically in a similar fashion to the HF group, as previously described.20,21
2.3 Isolation of mitochondria
Cardiac SSM and IFM were isolated from the LV using a reported protocol,17 with some modifications. Cardiac SSM were released by treating the minced muscle with collagenase 2 (Worthington Biochemical Corporation), 25 mg/g wet wt, for 10 min at 4°C in modified Chappel-Perry buffer (100 mM KCl, 50 mM Mops, 5 mM MgSO4·7H2O, 1 mM EGTA, 1 mM ATP, pH 7.4 at 4°C), followed by Potter-Elvehjem homogenization. The mitochondria enriched-supernatant (centrifugation for 10 min at ×584 g), was centrifuged at ×3000 g to pellet the SSM. The remaining myofibrillar pellet was treated with a combination of trypsin (5 mg/g wet wt, for 10 min at 4°C) and Polytron homogenization (10 000 r.p.m. for 2.5 s). The activity of the protease was attenuated with an equal volume of the modified Chappel-Perry buffer plus 0.2% defatted BSA, and the homogenate centrifuged at ×584 g for 10 min. The IFM were pelleted by centrifugation from the supernatant at ×3000 g for 10 min. Mitochondrial pellets were washed twice and finally suspended in KME (100 mM KCl, 50 mM Mops, and 0.5 mM EGTA, pH 7.4). Mitochondrial protein concentration was determined by the Lowry method using bovine serum albumin as a standard.22
2.4 Oxidative phosphorylation
Oxygen consumption was measured with a Clark-type electrode in a final volume of 0.5 mL of respiration buffer (80 mM KCl, 50 mM Mops, 1 mM EGTA, 5 mM KH2PO4, and 1 mg/mL defatted BSA, pH 7.4)23,24 at 30°C with glutamate (20 mM) without or with malate (5 mM), pyruvate (10 mM) plus malate (5 mM), succinate (20 mM) plus rotenone (3.75 µM), duroquinol (DHQ, 1 mM) plus rotenone, N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) plus ascorbate (1:10 mM) plus rotenone, as substrates. After depletion of endogenous substrates with 100 µM ADP (final concentration), State 3 respiration rate was measured in the presence of 200 µM ADP (ADP-dependent) and State 4 respiration was recorded after ADP consumption (ADP-limited). When mitochondria oxidize complex II, III, and IV substrates, respiratory state 3 was recorded with 100 µM ADP. Then sequential additions of 2 mM ADP and 200 µM of the uncoupler dinitrophenol (DNP) were made, as described.25 Respiratory control ratios (RCR, as State 3 divided by State 4) reflect the control of oxygen consumption by phosphorylation (‘coupling’). The ADP/O ratios (number of ADP molecules added for each oxygen atom consumed) are an index of the efficiency of oxidative phosphorylation. These indexes were calculated as described by Chance and Williams.26
2.5 Citrate synthase activity
Citrate synthase activity was measured using a diode array spectrophotometer at 412 nm.27
2.6 Mitochondrial electron transport chain complex activities
Samples of fresh SSM and IFM were treated with 10 mg cholate/mg mitochondrial protein, taken to a final volume of 1 mg/mL MSM-EDTA buffer supplemented with 1 µL/mL mammalian protease inhibitor cocktail. For the assay, the samples were diluted 1:10. For cytochrome c oxidase (COX) activity, 0.1 mg fresh intact mitochondria were suspended in KME buffer (100 mM KCl, 50 mM MOPS, and 0.5 mM EGTA, pH 7.4) and assayed with and without dodecyl β-D-maltoside.
ETC enzyme activities were measured spectrophotometrically as specific donor–acceptor oxidoreductase activities in 0.1 M phosphate buffer. Both donors and acceptors span specific regions of the ETC.23,28,29 Rotenone-sensitive NADH-cytochrome c reductase assesses complexes I and III. NADH coenzyme Q reductase was measured as the rotenone-sensitive oxidation of NADH with decylubiquinone as acceptor, and assesses only complex I. NADH ferricyanide reductase measures NADH dehydrogenase and mitochondrial outer membrane cytochrome b5 (rotenone-insensitive NADH-reductase). Antimycin A-sensitive succinate-cytochrome c reductase assesses complexes II and III. Complex II activity was measured as the thenoyltrifluoroacetone-sensitive reduction of 2,6-dicholorophenolindophenol with succinate as substrate, whereas total complex II was measured in the same manner but with duroquinone added as a source of exogenous coenzyme Q. Succinate dehydrogenase measures the first two subunits of complex II. Decylubiquinol-cytochrome c oxidoreductase was measured as the antimycin A-sensitive reductase to assess complex III. COX was measured as the oxidation of reduced cytochrome c and expressed as the first-order rate constant. Mitochondrial cytochrome contents (b, c1, aa3, c) were determined using the method of Williams.30
2.7 Polarographic assay of cytochrome c oxidase
Frozen-thawed mitochondria were suspended in buffer containing 20 mM KH2PO4 (pH 7.4) and 0.1 mM EDTA at a concentration of 1 mg mitochondrial protein per millilitre. The activity of COX was measured as azide-sensitive rate of oxygen consumption in the presence of 1 mM TMPD, 10 mM sodium ascorbate in potassium phosphate buffer at 30°C using the endogenous cytochrome c bound to the enzyme. A separate assay was performed in the presence of 8 µM exogenous cytochrome c. Because COX activity depends on the membrane cardiolipin content,31,32 the activity also was measured in the presence and absence of exogenous soybean asolectin, a source of phospholipids including cardiolipin.
2.8 Separation of mitochondrial supramolecular complexes by Blue Native PAGE electrophoresis
BN-PAGE was performed using the method of Schagger and Wittig.33 Mitochondrial proteins were extracted with digitonin (detergent:protein ratio of 6:1 g/g), combined with Coomasie blue G-250 dye (detergent/dye ratio of 8:1 g/g), and separated on 3–13% acrylamide–bisacrylamide gradient gels. Densitometric evaluation of the gels was carried out using Image J from the National Institutes of Health and Multi Gauge V3.0 from Fujifilm Life Science.
2.9 Statistical analysis
The results were analysed using the student’s two-tailed t-test. Data are reported as mean ± SEM. To be considered significant, P-value had to be less than 0.05.
3. Results
3.1 Haemodynamic findings
There was a progressive worsening in LV function from 1 to 13 weeks following the last microembolization, as evidenced by increases in the LV end-systolic volume (from 42 ± 1 to 47 ± 1 mL) and end-diastolic volume (from 64 ± 2 to 67 ± 2 mL), and a fall in ejection fraction (from 34 ± 1 to 28 ± 1) (P < 0.05 for all parameters).
3.2 Mitochondrial marker enzyme activities
Mitochondrial marker enzyme activities (citrate synthase and succinate dehydrogenase) in fresh heart homogenate, as indicators of mitochondrial content, were not changed in HF. The yields of heart SSM and IFM isolated from HF appear to be lower than those isolated from the control, but the alterations are not statistically significant (P-values are 0.227 for SSM and 0.065 for IFM). Also, no differences were found in the specific activity of mitochondrial marker enzymes in both SSM and IFM from control and HF dogs (Table 1).
Specific activities of mitochondrial marker enzymes in heart homogenate and isolated subsarcolemmal (SSM) and interfibrillar (IFM) mitochondria
| Control | Heart failure | |
|---|---|---|
| Heart homogenate | ||
| Citrate synthase (U/g wet wt) | 81.0 ± 5.0 | 84.0 ± 5.4 |
| Succinate dehydrogenase, (U/g wet wt) | 5.0 ± 0.3 | 5.0 ± 0.8 |
| Isolated mitochondria | ||
| Citrate synthase, (mU/mg protein) | ||
| SSM | 1975 ± 139 | 1902 ± 297 |
| IFM | 2136 ± 166 | 2088 ± 258 |
| Succinate dehydrogenase, (mU/mg protein) | ||
| SSM | 261 ± 40 | 251 ± 21 |
| IFM | 268 ± 45 | 261 ± 34 |
| Mitochondrial yield (mg/g wet wt) | ||
| SSM | 10.5 ± 0.7 | 7.6 ± 1.8 |
| IFM | 10.6 ± 1.3 | 5.4 ± 1.2 |
| Control | Heart failure | |
|---|---|---|
| Heart homogenate | ||
| Citrate synthase (U/g wet wt) | 81.0 ± 5.0 | 84.0 ± 5.4 |
| Succinate dehydrogenase, (U/g wet wt) | 5.0 ± 0.3 | 5.0 ± 0.8 |
| Isolated mitochondria | ||
| Citrate synthase, (mU/mg protein) | ||
| SSM | 1975 ± 139 | 1902 ± 297 |
| IFM | 2136 ± 166 | 2088 ± 258 |
| Succinate dehydrogenase, (mU/mg protein) | ||
| SSM | 261 ± 40 | 251 ± 21 |
| IFM | 268 ± 45 | 261 ± 34 |
| Mitochondrial yield (mg/g wet wt) | ||
| SSM | 10.5 ± 0.7 | 7.6 ± 1.8 |
| IFM | 10.6 ± 1.3 | 5.4 ± 1.2 |
n = 5 for control and n = 5 for heart failure. Mean ± SEM.
Specific activities of mitochondrial marker enzymes in heart homogenate and isolated subsarcolemmal (SSM) and interfibrillar (IFM) mitochondria
| Control | Heart failure | |
|---|---|---|
| Heart homogenate | ||
| Citrate synthase (U/g wet wt) | 81.0 ± 5.0 | 84.0 ± 5.4 |
| Succinate dehydrogenase, (U/g wet wt) | 5.0 ± 0.3 | 5.0 ± 0.8 |
| Isolated mitochondria | ||
| Citrate synthase, (mU/mg protein) | ||
| SSM | 1975 ± 139 | 1902 ± 297 |
| IFM | 2136 ± 166 | 2088 ± 258 |
| Succinate dehydrogenase, (mU/mg protein) | ||
| SSM | 261 ± 40 | 251 ± 21 |
| IFM | 268 ± 45 | 261 ± 34 |
| Mitochondrial yield (mg/g wet wt) | ||
| SSM | 10.5 ± 0.7 | 7.6 ± 1.8 |
| IFM | 10.6 ± 1.3 | 5.4 ± 1.2 |
| Control | Heart failure | |
|---|---|---|
| Heart homogenate | ||
| Citrate synthase (U/g wet wt) | 81.0 ± 5.0 | 84.0 ± 5.4 |
| Succinate dehydrogenase, (U/g wet wt) | 5.0 ± 0.3 | 5.0 ± 0.8 |
| Isolated mitochondria | ||
| Citrate synthase, (mU/mg protein) | ||
| SSM | 1975 ± 139 | 1902 ± 297 |
| IFM | 2136 ± 166 | 2088 ± 258 |
| Succinate dehydrogenase, (mU/mg protein) | ||
| SSM | 261 ± 40 | 251 ± 21 |
| IFM | 268 ± 45 | 261 ± 34 |
| Mitochondrial yield (mg/g wet wt) | ||
| SSM | 10.5 ± 0.7 | 7.6 ± 1.8 |
| IFM | 10.6 ± 1.3 | 5.4 ± 1.2 |
n = 5 for control and n = 5 for heart failure. Mean ± SEM.
3.3 Oxidative phosphorylation
Glutamate oxidation (Table 2) reflects mitochondrial glutamate uptake, glutamate dehydrogenase, the activity of the ETC, and the phosphorylation process. Both SSM and IFM isolated from HF have state 3 respiratory rates that are approximately 50% of those from control hearts.
Oxidative properties of subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM) isolated from heart of control and heart failure dogs
| Control | Heart failure | |
|---|---|---|
| SSM | ||
| State 3 | 514 ± 41 | 214 ± 33* |
| State 4 | 15 ± 5 | 15 ± 3 |
| RCR | 34 ± 7 | 18 ± 4 |
| ADP/O | 2.77 ± 0.09 | 2.69 ± 0.02 |
| Maximal ADP | 619 ± 41 | 191 ± 22* |
| DNP | 608 ± 32 | 191 ± 38* |
| IFM | ||
| State 3 | 472 ± 26 | 223 ± 12* |
| State 4 | 20 ± 6 | 16 ± 4 |
| RCR | 39 ± 14 | 17 ± 3 |
| ADP/O | 2.78 ± 0.15 | 2.85 ± 0.15 |
| Maximal ADP | 529 ± 16 | 221 ± 17* |
| DNP | 535 ± 10 | 206 ± 19* |
| Control | Heart failure | |
|---|---|---|
| SSM | ||
| State 3 | 514 ± 41 | 214 ± 33* |
| State 4 | 15 ± 5 | 15 ± 3 |
| RCR | 34 ± 7 | 18 ± 4 |
| ADP/O | 2.77 ± 0.09 | 2.69 ± 0.02 |
| Maximal ADP | 619 ± 41 | 191 ± 22* |
| DNP | 608 ± 32 | 191 ± 38* |
| IFM | ||
| State 3 | 472 ± 26 | 223 ± 12* |
| State 4 | 20 ± 6 | 16 ± 4 |
| RCR | 39 ± 14 | 17 ± 3 |
| ADP/O | 2.78 ± 0.15 | 2.85 ± 0.15 |
| Maximal ADP | 529 ± 16 | 221 ± 17* |
| DNP | 535 ± 10 | 206 ± 19* |
Glutamate as substrate. RCR, respiratory control ratio (state 3/state 4); ADP/O, ADP to atomic oxygen phosphorylation ratio; Maximal ADP, state 3 with saturating 2 mM ADP; DNP, dinitrophenol. Respiratory rates are expressed as nA O/min/mg mitochondrial protein. n= 5 for control and n = 5 for HF. Mean ± SEM.
*P < 0.05.
Oxidative properties of subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM) isolated from heart of control and heart failure dogs
| Control | Heart failure | |
|---|---|---|
| SSM | ||
| State 3 | 514 ± 41 | 214 ± 33* |
| State 4 | 15 ± 5 | 15 ± 3 |
| RCR | 34 ± 7 | 18 ± 4 |
| ADP/O | 2.77 ± 0.09 | 2.69 ± 0.02 |
| Maximal ADP | 619 ± 41 | 191 ± 22* |
| DNP | 608 ± 32 | 191 ± 38* |
| IFM | ||
| State 3 | 472 ± 26 | 223 ± 12* |
| State 4 | 20 ± 6 | 16 ± 4 |
| RCR | 39 ± 14 | 17 ± 3 |
| ADP/O | 2.78 ± 0.15 | 2.85 ± 0.15 |
| Maximal ADP | 529 ± 16 | 221 ± 17* |
| DNP | 535 ± 10 | 206 ± 19* |
| Control | Heart failure | |
|---|---|---|
| SSM | ||
| State 3 | 514 ± 41 | 214 ± 33* |
| State 4 | 15 ± 5 | 15 ± 3 |
| RCR | 34 ± 7 | 18 ± 4 |
| ADP/O | 2.77 ± 0.09 | 2.69 ± 0.02 |
| Maximal ADP | 619 ± 41 | 191 ± 22* |
| DNP | 608 ± 32 | 191 ± 38* |
| IFM | ||
| State 3 | 472 ± 26 | 223 ± 12* |
| State 4 | 20 ± 6 | 16 ± 4 |
| RCR | 39 ± 14 | 17 ± 3 |
| ADP/O | 2.78 ± 0.15 | 2.85 ± 0.15 |
| Maximal ADP | 529 ± 16 | 221 ± 17* |
| DNP | 535 ± 10 | 206 ± 19* |
Glutamate as substrate. RCR, respiratory control ratio (state 3/state 4); ADP/O, ADP to atomic oxygen phosphorylation ratio; Maximal ADP, state 3 with saturating 2 mM ADP; DNP, dinitrophenol. Respiratory rates are expressed as nA O/min/mg mitochondrial protein. n= 5 for control and n = 5 for HF. Mean ± SEM.
*P < 0.05.
The RCRs (state 3/state 4) in heart SSM and IFM from control dogs are 34 ± 7 and 39 ± 14, respectively, with glutamate as substrate. These values demonstrate that both heart SSM and IFM isolated from control dogs are highly coupled. These RCRs for dog heart mitochondria are higher than those reported by our group for rat heart mitochondria oxidizing glutamate,17 but are similar to those reported (from 33 ± 7.5 to 39.6 ± 1.6) in the literature for the combined population (SSM + IFM) of mitochondria isolated from chicken heart.34 The RCR values were slightly decreased in HF due exclusively to the decline in state 3 respiratory rates, but the values did not reach statistical significance. State 4 respiratory rates were 15 ± 5 nA O/min/mg in control dog heart SSM and 20 ± 6 nA O/min/mg in control dog heart IFM oxidizing glutamate, and these values did not change in mitochondria isolated from HF (15 ± 3 nA O/min/mg in SSM and 16 ± 4 nA O/min/mg in IFM). These state 4 respiratory rates are similar to those measured in rat heart mitochondria.17 Thus, the notable RCRs reflect the high state 3 rates of oxidation rather than a decrease in state 4 in dog heart mitochondria.
Also, the ADP/O ratios were not changed in either SSM or IFM isolated from HF compared with the control hearts. These data revealed that, despite the severe decline in state 3, the coupling abilities of both populations of heart mitochondria are preserved in HF. The decreased respiratory rates were not relieved by the addition of either saturating concentration of ADP or by the uncoupler DNP, showing that the mitochondrial defect does not reside in the phosphorylation apparatus.
In order to dissect the oxidative phosphorylation into functional components, we also employed substrates that use different mitochondrial transport systems and dehydrogenases, the tricarboxylic acid cycle, and that donate their reducing equivalents to selective sites in the ETC.
Figure 1 shows the respiratory properties of heart SSM and IFM in the presence of complex I, II, III, and IV substrates. The combination of glutamate + malate investigates the aspartate shuttle, whereas pyruvate + malate assesses the monocarboxylate and dicarboxylate transporters and pyruvate dehydrogenase. Both substrate combinations produce NADH, which donates electrons to complex I. Heart SSM and IFM isolated from HF had approximately 45% state 3 respiratory rates compared with control mitochondria, irrespective of complex I substrates processing. CPT-I-dependent and -independent lipid substrates (complex I and complex III electron donors, via electron transfer flavoprotein-ETF and ETF-Q reductase) also were oxidized at significantly lower rates (see Supplementary material online, Figure S1).
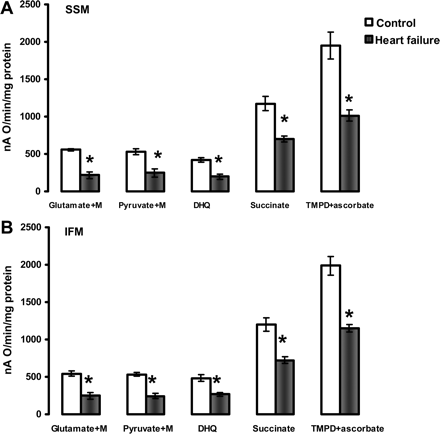
State 3 respiratory rates of heart subsarcolemmal mitochondria (A) and interfibrillar mitochondria (B). State 3 was induced by 200 µM ADP when mitochondria oxidize complex I substrates (Glutamate+Malate and Pyruvate+Malate), and 100 µM ADP when mitochondria oxidize complexes II (Succinate+rotenone), III (Duroquinol, DHQ+rotenone), and IV via cytochrome c (TMPD ascorbate+rotenone). M, malate. *P < 0.05 control (n = 5) vs. heart failure (n = 5). Mean ± SEM.
Succinate donates electrons to FAD in complex II, and yields significantly lower respiratory state 3 rates in both cardiac SSM and IFM isolated from HF compared with the control. The respiratory state 3 rates with duroquinol, as a complex III electron donor, were significantly lower in both SSM and IFM isolated from HF compared with the control hearts. State 3 respiratory rates with the cytochrome c electron donor TMPD-ascorbate also were significantly decreased by HF in both SSM and IFM.
A definitive localization of the defect in the oxidative phosphorylation process is provided by measuring the uncoupled respiratory rates. The lack of improvement of respiratory rates upon collapsing of mitochondrial potential with the protonophore DNP with all substrates tested showed that neither the amount nor the activity of the components of the phosphorylation apparatus (adenine nucleotide translocase-ANT isoforms, phosphate transporter, ATP synthase) were the sites of the defect in both cardiac SSM and IFM isolated from HF (Figure 2). Moreover, respiratory state 3 rates were not improved by the addition of saturating concentrations of ADP (see Supplementary material online, Figure S2) showing that the defect was localized in mitochondrial structures situated upstream from ANT in the respiratory chain.
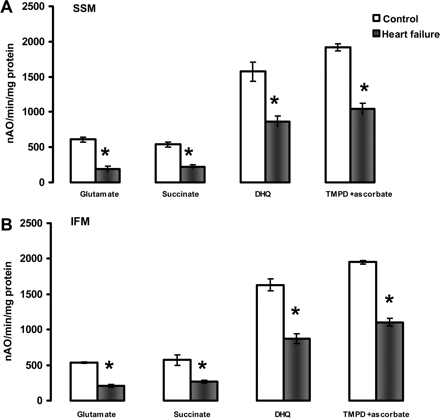
Uncoupled (200 µM dinitrophenol) respiratory rates of heart subsarcolemmal mitochondria (A) and interfibrillar mitochondria (B). Substrates that donate reducing equivalents to complexes I (Glutamate), II (Succinate+rotenone), III (Duroquinol, DHQ+rotenone), and IV via cytochrome c (TMPD ascorbate+rotenone). *P < 0.05 control (n = 5) vs. heart failure (n = 5). Mean ± SEM.
3.4 The activity of mitochondrial electron transport chain complexes
The individual activities of the mitochondrial ETC enzymes were not affected (Figure 3). In addition, COX activity, measured under isosmotic conditions to assess outer membrane leakage, was 3.2 ± 1 and 2.5 ± 0.5% of the total activity in control SSM and IFM, respectively, and was not modified in HF. These data show that the outer membrane is essentially intact and is preserved in HF in both heart mitochondrial populations.
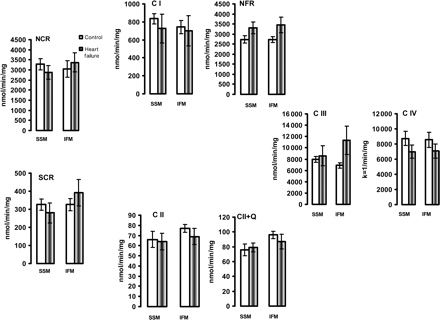
The activity of electron transport chain enzymes in isolated heart subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM). The enzyme activity was measured in detergent-solubilized, freshly isolated SSM and IFM from control (n = 5) and heart failure (n = 5) dogs, and expressed as nmol/min/mg mitochondrial protein or as the first-order rate constant (k = 1/min/mg mitochondrial protein) for complex IV. NCR, rotenone sensitive NADH-Cytochrome c Reductase; C I, complex I; NFR, NADH-Ferricyanide Reductase; SCR, antimycin A-sensitive Succinate-Cytochrome c Reductase. C II, TTFA-sensitive complex II; C II+Q, TTFA-sensitive Complex II with exogenous coenzyme Q analogue; C III, complex III; C IV, complex IV. Mean ± SEM.
Also, when measured in fresh heart muscle homogenate, the combined activities of complexes I/III (rotenone-sensitive NADH cytochrome c reductase, NCR) and complexes II/III (antimycin-sensitive succinate cytochrome c reductase, SCR), as well as the activity of decylubiquinol-cytochrome c oxidoreductase (complex III) and COX (complex IV) were unchanged, consistent with the activities of these enzymes in isolated mitochondria (Supplementary material online, Figure S3).
3.5 Polarographic investigation of cytochrome c oxidase
The normal activity observed with the spectrophotometric assay performed in the presence of exogenously added cytochrome c assesses predominantly the activity of the low-affinity high-turnover binding site of cytochrome c to COX (regulatory site).35 The polarographic investigation of COX in the absence of exogenously added cytochrome c measures the velocity of oxygen consumption under conditions whereby TMPD-ascorbate maintains the cytochrome c bound to COX to the high affinity–low turnover site (HA–LT, catalytic site) in the reduced state. COX activity measured polarographically was not altered by HF in both SSM and IFM in the absence of exogenously added cytochrome c, and was increased to the same extent in both types of control and HF heart mitochondria by the addition of exogenous cytochrome c (Figure 4). These results show that neither the individual COX activity nor the reducible (functional) cytochrome bound to COX to the HA–LT site are altered in HF. The effect of asolectin (as a source of exogenous phospholipids) on the increase in COX activity was similar in mitochondria isolated from both control and HF dogs (Figure 4).
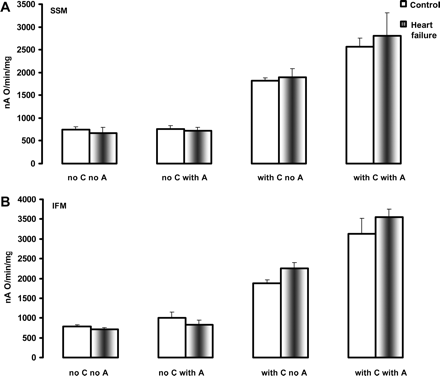
The activity of cytochrome c oxidase in heart subsarcolemmal mitochondria (A) and interfibrillar mitochondria (B) determined polarographically. C, cytochrome c. A, asolectin. Mean ± SEM.
3.6 Cytochrome content
The amount of cytochrome aa3 reducible haem, as the main catalytic subunit of COX, as well as the amount of the reducible haem in cytochromes b and c was unchanged in both SSM and IFM isolated from HF. The amount of cytochrome c1, contained in complex III, was significantly increased in IFM isolated from HF compared with the control (Figure 5).
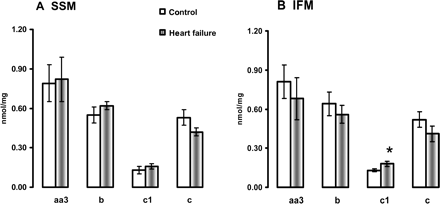
Cytochrome content in heart subsarcolemmal mitochondria (A) and interfibrillar mitochondria (B). *P < 0.05 control vs. HF. Mean ± SEM.
3.7 Supramolecular complex assemblies and individual electron transport chain complexes
The results presented in Figures 6 and 7 provide data showing that HF causes a decrease in the amount of the major mitochondrial supercomplex (respirasome I/III2/IV) in both heart SSM and IFM. One-dimensional BN-PAGE separation of mitochondrial supercomplexes extracted with digitonin is shown in Figures 6A and 7A. Since the amount of complex V is not changed (Figures 6B and 7B) and complex V is not a component of this supercomplex,33,36,37 the results are further expressed as the intensity of the supercomplex bands normalized to the intensity of complex V band. Figures 6C and 7C revealed a lower amount of the supercomplex I/III2/IV (described by Schagger33,36,37) in both types of heart mitochondria in HF. In contrast, the amounts of free complexes I and III were increased in HF in SSM with free complex IV unchanged. Also, free complexes I, III, and IV migrate at a similar molecular weight in both control and HF, indicating that they remain stable when not part of the supercomplex and that they have subunit integrity (see Supplementary material online, Figure S4).
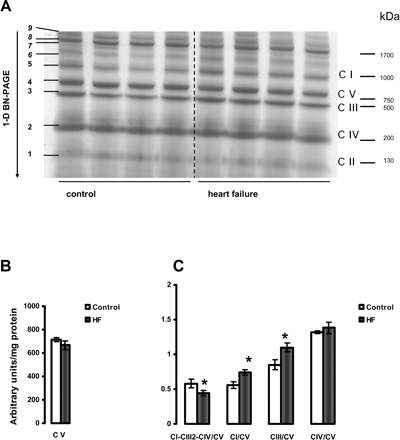
Separation of supramolecular assemblies of mitochondrial oxidative phosphorylation complexes by 1D BN-PAGE in subsarcolemmal mitochondria (SSM) of control and heart failure hearts. (A) Dog heart SSM solubilized with digitonin using a 6:1 digitonin/protein ratio (g/g) and separated by one-dimensional BN-PAGE. C I, C II, C III, C IV, and C V (right) indicate mitochondrial complexes I–V and correspond to bands 1–5 (left). Bands 6–9 (left) indicate supermolecular complexes. Band 8 indicates supercomplex CI-CIII2-CIV. (B) Complex V (C V). (C) The density of the band corresponding to supercomplex CI-CIII2-CIV (arrows) and of the bands corresponding to complexes I, III, and IV were normalized to the density of complex V band. *P < 0.05 control vs. HF. Mean ± SEM. One-dimensional BN-PAGE, one-dimensional blue native PAGE.
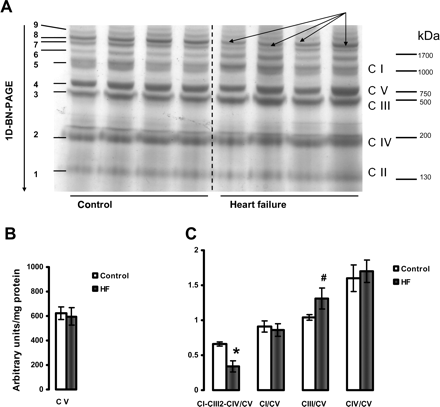
Separation of supramolecular assemblies of mitochondrial oxidative phosphorylation complexes by one-dimensional BN-PAGE in interfibrillar mitochondria (IFM) of control and heart failure hearts. (A) Dog heart IFM solubilized with digitonin using a 6:1 digitonin/protein ratio (g/g) and separated by 1D BN-PAGE. C I, C II, C III, C IV, and C V (right) indicate mitochondrial complexes I–V and correspond to bands 1–5 (left). Bands 6–9 (left) indicate supermolecular complexes. Band 8 (arrows) indicates supercomplex CI-CIII2-CIV. (B) Complex V (C V). (C) The density of the band corresponding to supercomplex CI-CIII2-CIV (arrows) and of the bands corresponding to complexes I, III, and IV were normalized to the density of complex V band. *P < 0.05 control vs. HF. #P = 0.07 control vs. HF. Mean ± SEM. One-dimensional BN-PAGE, one-dimensional blue native PAGE.
4. Discussion
This study documents that there is a severe decrease in oxidative phosphorylation of both populations of heart mitochondria (SSM and IFM) isolated from moderate severity HF because of a decrease in the content of a major ETC supercomplex (respirasome). The content of mitochondria in the myocardium was unchanged in HF. Mitochondria were intact with little or no outer membrane leakiness. Heart mitochondria showed a dramatic decrease in the ADP-stimulated respiration that was not relieved by an uncoupler. RCR values are unchanged, indicating that mitochondria are well-coupled. Also, the ADP/O ratio was normal, indicating that HF does not affect the efficiency of phosphorylation. No change in the individual activity of the ETC complexes was found in HF. Therefore, the mitochondrial defect resides in the assembly and function of the mitochondrial respirasome rather than in individual complexes.
The functional defect can only be uncovered by a comprehensive evaluation of mitochondria. Our approach involves the measurement of oxidative phosphorylation, complemented with ETC complex activities, mobile electron carriers, and coupled with the analysis of the organization of the oxidative phosphorylation complexes into supercomplexes.
Oxidative phosphorylation, performed on freshly isolated mitochondria with a comprehensive number of substrates, assesses the integrated function of membrane transport, dehydrogenase activity, and electron transport coupled to ATP synthesis.25 The lack of improvement of oxidative phosphorylation rates by collapsing the mitochondrial membrane potential with an uncoupler shows that the defect is not localized in any structure of the phosphorylation system (ANT, ATP synthase, phosphate transporter), but rather in electron transport. These data extend previous studies in the literature.9,16 However, a great variability of mitochondrial ETC defects is described in acquired cardiomyopathies4,6,38–40 without a consistent trend for depressed activity or expression. In this study, the dramatic drop in oxidation of all substrates was not supported by defects in the individual activity of any ETC complex. COX activity, total and COX-bound reducible (functional) form of cytochrome c, and both catalytic and regulatory COX cytochrome c binding sites also are normal in HF.
Complexes of the mitochondrial oxidative phosphorylation system assemble into supramolecular structures called respiratory supercomplexes, as revealed by (i) kinetic studies using flux control analysis,41 (ii) single particle electron microscopy,42,43 (iii) studies that show the instability of human respirasome in patients with genetic mitochondrial disorders,36 (iv) studies on the stability of oxidative phosphorylation complexes with induced point mutations in a specific complex,44 and (v) separation by BN-PAGE procedures, which yield assemblies of oxidative phosphorylation enzymes rather than individual complexes. The supramolecular organization of the oxidative phosphorylation system is strongly conserved from fungal,45 plant,46 beef,37,42 to human37 mitochondria. Supercomplexes consisting of complexes I/dimer III/one to four copies of IV (I/III2/IV1-4) were proposed to be called respirasomes,36 because they autonomously can carry out respiration in the presence of ubiquinone and cytochrome c. In yeast complex IV assemblies with complex III and does not occur significantly in free form.37 The I/III2/IV1–4 supercomplexes represent the major forms of respirasomes in mammalian mitochondria. Also, in mammalian mitochondria, almost all complex I is assembled into respirasomes.37 Moreover, we have found complex I only in respirasomes, but not in the free monomeric form in normal human skeletal muscle (unpublished observation).
We report that the amount of the supramolecular complex I/III2/IV is decreased in HF in both heart mitochondrial populations. In contrast, the amounts of free monomeric complexes I and III are significantly increased in heart SSM isolated from HF. However, the total amounts of complexes I and III extracted by dodecyl β-D-maltoside are unchanged. Therefore, there is a decreased incorporation of complexes I and III within I/III2/IV respirasome in SSM.
By facilitating the activity of catalytic subunits, supercomplexes represent regulatory units of mitochondrial oxidative phosphorylation.47 We propose that the decrease in supramolecular organization of the oxidative phosphorylation leading to loss of the functional advantage of this respirasome organization, such as channelling of electrons and substrates,48 causes the severe decrease in the integrative mitochondrial function. Our proposal is supported by studies on supramolecular assembly of mitochondrial oxidative phosphorylation in humans with mitochondrial disorders. For example, humans harbouring a genetic defect in cardiolipin remodelling (Barth syndrome) have unstable oxidative phosphorylation supercomplexes, associated with mitochondrial morphological abnormalities,49 decreased mitochondrial potential,50 and decreased activity of the ETC complexes.51 Also, after screening a large number of mitochondrial specimens from muscle biopsies of patients with mitochondrial disorders, Schagger36 reported deficiencies in the assembly of complexes I and IV.
The 50% decrease in mitochondrial oxidative phosphorylation with normal individual activity of the ETC complexes was accompanied by an approximately 33 and 50% decrease in the amount of respirasome in SSM and IFM, respectively. We propose that the severe decrease in oxidative phosphorylation is dependent upon the formation of functional supercomplexes. This idea is supported by recent studies in hybrid cells harbouring mtDNA mutations within COX I and cytochrome b genes that experience both a collapse in oxidative phosphorylation and complete absence of fully assembled functional supercomplexes. The decrease in the proportion of these mtDNA mutations leads to increase in the complex III- and IV-containing supercomplexes rather than their free form, and a complete recovery of oxidative phosphorylation. The authors concluded that the amount and activity of individual complexes are not rate-limiting for mitochondrial respiration, and it is the ability of the ETC to organize in supercomplexes that sets the threshold for the phenotypic expression of the mtDNA defects.52 The amount of free complexes I and III was increased in SSM isolated from HF hearts. In IFM, there is a trend toward higher free complex III amount. These free complexes might represent a functional reserve and be in a dynamic equilibrium with those organized in supercomplexes, as suggested by the semi-solid-state model of the organization of oxidative phosphorylation.52
The present study demonstrates that both populations of heart mitochondria are equally affected during this model of microembolism-induced HF of moderate severity. This conclusion is supported by both the oxidative phosphorylation data and the amount of supercomplexes.
In summary, the current study documents a dramatic decrease in oxidative phosphorylation of heart mitochondria isolated from moderately severe HF. The defect is caused by a decrease in the content of the major mitochondrial supramolecular complex (respirasome). Our data reveal a new mitochondrial disorder in HF that fits in the category of mitochondrial cytopathies.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
Funding for this work was provided by the National Heart, Lung and Blood Institute, Program Project Grant PO1 (HL-074237).
Acknowledgements
We are grateful to Hazel Huang for excellent technical assistance with regard to dog surgery. We also thank Hiral Patel, Amy Foster, and Kalpena Patel for performing the ETC analysis. We acknowledge Dr Bernard Tandler for editorial assistance.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}