





Abstract
The aim of this study was to take a combination of animal and cell culture approaches to examine the individual responses of vascular cells to varying inflammatory factors in order to gain insights on the mechanism(s) by which poly(ADP-ribose) polymerase (PARP) inhibition promotes factors of plaque stability.
Apolipoprotein (ApoE−/−) mice fed a high-fat diet were used as a model of atherosclerosis. Primary endothelial cells, smooth muscle cells (SMCs), and ex-vivo generated foam cells (FCs) were used in our in vitro studies. PARP inhibition significantly decreased the markers of oxidative stress and caspase-3 activation and increased smooth muscle actin within plaques from ApoE−/− mice fed a high-fat diet. PARP inhibition protected against apoptosis and/or necrosis in SMCs and endothelial cells in response to H2O2 or tumour necrosis factor (TNF). Remarkably, PARP inhibition in FCs resulted in significant sensitization to 7-ketocholesterol (7-KC) by increasing cellular-toxic-free cholesterol, potentially through a down-regulation of acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) expression. 7-KC induced necrosis exclusively in endothelial cells, which was, surprisingly, unaffected by PARP inhibition indicating that PARP inhibition does not prevent all forms of necrotic cell death. In SMCs, PARP-1 inhibition by gene deletion conferred protection against 7-KC or TNF, potentially by reducing caspase-3-like activation, preventing induction of c-Jun N-terminal protein kinase phosphorylation, and inducing extracellular signal-regulated kinase phosphorylation independently of PARP classical enzymatic activity.
These data present PARP-1 as an important player in the death of cells constituting atherosclerotic plaques contributing to plaque dynamics. PARP inhibition may be a protective, a neutral, or a sensitizing factor. Additionally, PARP-1 may be a novel factor that can alter lipid metabolism. These novel functions of PARP not only challenge the current understanding of the role of the enzyme in cell death but also provide insights on the intricate contribution of PARP in cellular responses to predominant inflammatory factors within atherosclerotic plaques, presenting additional evidence for the viability of PARP inhibition as a therapeutic strategy for atherosclerosis.
1. Introduction
Atherosclerosis is a progressive inflammatory disease that involves medium and large arteries and is characterized by the progressive accumulation of intra- and extracellular lipids, smooth muscle cells (SMCs), macrophages, and connective tissue.1,2 Inflammation plays a key role in the pathogenesis of atherosclerosis and is accompanied by modified low-density lipoprotein (LDL), hypertension, and free radicals.2 Multiple risk factors induce the generation of reactive oxygen and nitrogen species, which enhance oxidative stress in vascular cells by activating a number of inflammatory cytokines (reviewed in2). Atherosclerotic plaque vulnerability is increasingly becoming a major target for drug design and therapeutic strategies.
PARP is a family of proteins of which PARP-1 is an important member. PARP-1 is a DNA repair-associated nuclear enzyme, which is activated in response to DNA damage. PARP-1 activation catalyses the covalent coupling of branched chains of ADP-ribose units to various nuclear proteins such as histones and PARP-1 itself. Excessive PARP-1 activation following DNA damage upon exposure to pro-inflammatory agents leads to acute depletion of the metabolic intermediates nicotinamide adenine dinucleotide (NAD) and ATP. This activation leads to cell death because of energy deprivation, which ultimately participates in the overall process of inflammation.3 Though initially implicated in necrotic cell death,4 PARP-1 has been found by our laboratory to be an active agent in the early stages of the apoptotic process.5
Recently, we have shown that PARP inhibition interferes with plaque development and may promote factors of plaque stability including a reduction in inflammatory factors and cellular changes related to plaque dynamics.6,7 PARP inhibition was shown to reduce both plaque size and number, but more importantly, PARP inhibition increased SMCs content, decreased collagen degradation, and increased tissue inhibitor of metalloproteinase-2 expression, all of which are considered as factors of plaque stability. Given the complexity of atherosclerotic plaques, the present study was designed to decipher the mechanism(s) by which PARP inhibition alters the dynamics of atherosclerotic plaques using both the animal model of the disease and an ex vivo system of cell culture wherein plaque was virtually dissected. We wished to test the hypothesis that PARP-1 plays an important role in the fate of cells constituting atherosclerotic plaques in response to different inflammatory factors and that its inhibition provides conditions conducive to cell survival ultimately contributing to plaque stability. The results of the present study indicate that PARP inhibition decreases the inflammatory response, reduces oxidative tissue damage, and selectively promotes FC death while protecting endothelial and SMCs from injury by H2O2, oxidized cholesterol, or tumour necrosis factor (TNF). These features of PARP inhibition are highly desirable for a therapeutic strategy to stabilize or reduce atherosclerosis plaques.
2. Methods
2.1 Animals and diet protocol
C57BL/6 wild-type (WT) (Jackson Laboratory, Bar Harbor, ME, USA), Apolipoprotein (ApoE−/−) (Jackson Laboratory) and PARP-1−/− mice were housed and bred in a pathogen-free animal care facility at LSUHSC (New Orleans, LA, USA) and allowed full access to laboratory rodent chow and water. Principles of laboratory animal care (NIH publication No. 86-23, revised 1985) were followed and experimental protocols were approved by the LSUHSC Animal Care and Use Committee. The C57BL/6 PARP-1−/− mice were generated as described.8 Six-week-old ApoE−/− mice received regular chow or a high-fat diet (Harlan Teklad, Madison, WI, USA) containing 21% fat by weight (0.15% cholesterol) and were divided into groups receiving i.p. injections of 3 mg/kg of the PARP inhibitor thieno[2,3-c]isoquinolin-5-one (TIQ-A) (Sigma-Aldrich, St Louis, MO, USA)9 two times per week or vehicle alone.
2.2 Histology, immunohistochemistry, and quantitation of immunoreactivity
Aortas were processed for histology or immunohistochemistry using standard protocols, with antibodies to murine smooth muscle actin (SMA) (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), to nitro-tyrosine (Upstate, Lake Placid, NY, USA), to 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG) (Oxis International, Foster City, CA, USA), or to active caspase-3 (Cell Signaling, Danvers, MA, USA). Immunoreactivity was assessed in captured images of immunostained sections analysed with the use of Image-Pro Plus (Silver Spring, MD, USA) as described previously.6
2.3 Cell Culture, viability, intracellular quantitation of NAD, and FACS analysis
Isolation of macrophages and SMCs were from WT and PARP-1−/− mice were conducted as described.6 SMCs were used at passages four to six. Human umbilical vein endothelial cells (HUVEC) cells were purchased from Cascade Biologies Inc. (Portland, OR, USA). PARP inhibition in HUVEC was achieved pharmacologically by treatment with TIQ-A because isolation of endothelial cells from mice proved difficult. For FC preparation, macrophages were serum-starved for 2 h, after which 5 µg/mL acetylated LDL (Biomedical Technologies, Stoughton, MA, USA) was added. After 24 h, most of the macrophages changed their phenotype into FCs as determined by oil red O staining. The following inflammatory agents were utilized: H2O2, a representative oxidant; TNF, a cytokine; and 7-ketocholeserol (7-KC), a representative oxidized cholesterol.10 Cell viability and fluorescence activated cell sorting (FACS) analysis were conducted as described6 while intracellular amounts of NAD were measured as described.5
2.4 Assay of caspase-3 activity
Caspase-3 activity was measured essentially as described.5 In brief, cell extracts (25 µg of protein) were incubated for 10 min at 37°C with 40 µM N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (DEVD-AMC) peptide substrate in a total volume of 200 µL. The fluorescence of free AMC was monitored continuously over 10 min with a fluorometer at excitation and emission wavelengths of 360 and 460 nm, respectively.
2.5 Western immunoblot analysis and real-time RT-PCR
All these methods were performed using standard protocols. Real-time PCR was performed using cDNA generated using RNA isolated from control or 7-KC-treated wild type or PARP-1−/− FCs using a forward primer (5′-GTATTTGCAACGGAG-GAGGA-3′) and a reverse primer (5′-GCCCAAGGGAGTCACATTTA-3′). The β-actin primers were described in.6 Amplification, detection, and data analysis were performed with the iCycler real-time PCR system (Bio-Rad Laboratories, Hercules, CA, USA).
2.6 Immunofluorescence microscopy
Cells were fixed, permeabilized, and stained with a rabbit antibody to mouse Acyl-CoA:cholesterol acyltransferase (ACAT-1) and counterstained with DAPI essentially as described.5 The stained cells were then examined with an Olympus fluorescence microscope.
2.7 Total and free cholesterol determination
Cells were subjected to chloroform/methanol extraction to isolate cellular lipids and proteins. Total and free cholesterol were determined using a kit from Biovision Inc. (Mountain View, CA, USA) per manufacturer instructions.
2.8 Statistical analysis
All data are expressed as mean ± SD of values from at least six mice per group or from four to six replicates of the same treatment of the cultured cells. PRISM software (GraphPad, San Diego, CA, USA) was used to analyse the differences between experimental groups by 1-way ANOVA followed by the Dunnett multiple comparison test. Pvalues of <0.05 were considered significant.
3. Results
3.1 PARP inhibition selectively favours stability through alterations of plaque composition and structure by reducing oxidative DNA damage and protein nitration
Figure 1A and B show the obvious cellular and structural changes caused by TIQ-A treatment as plaques of mice that were treated with the drug show mostly well-defined SMCs as well as distinctly atrophied and ‘unhealthy’ FC. To examine the mechanism(s) by which PARP inhibition promotes factors of plaques stability, we examined the molecular aspects of the plaque that were altered in the high-fat diet-induced atherosclerosis mouse model. By concentrating on TIQ-A dosage (3 mg/kg, twice a week) that does not significantly affect plaque size but does promote factors of plaque stability, it allowed us to focus, specifically, on the effect(s) of several factors that are known to contribute to plaque dynamics namely oxidative stress. PARP inhibition resulted in a significant decreased in 8-oxo-dG and nitrotyrosine, which constitutes important markers of oxidative tissue injury (Figure 1C–D). These results suggest that PARP inhibition increases factors of plaque stability potentially by modulating oxidative damage associated with plaque disruption.
![PARP inhibition causes structural changes and reduces markers of oxidative stress within atherosclerotic plaques of high-fat-diet-fed ApoE−/− mice. (A) H&E stain of aortas from ApoE−/− high-fat (H-F) diet or high-fat with thieno[2,3-c]isoquinolin-5-one injections (3 mg/kg) twice a week (H-F+TIQ-A) for 12 weeks; FC, foam cells. Immunohistochemistry analysis of plaques from high-fat and high-fat with thieno[2,3-c]isoquinolin-5-one animal groups with antibodies to SMA (B), nitro-tyrosine (C), or to 8-oxo-dG (D); the rightmost panels are quantifications of immunoreactivity using Image-Pro Plus software and expressed as fold change from plaques of high-fat-diet-fed ApoE−/− mice. *P < 0.01). Bars: 3 µm.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/78/3/10.1093/cvr/cvn018/2/m_cvn01801.gif?Expires=1716503058&Signature=IjBJmfrixCYTJQ1b-IvPgRBrG7zIxtdxygCUrazcIgfOb9YImYlOuyJxHkKfJ-SQGW~aMHPjRN5KBM1oY2z7XSSs0yHlUAISg3owWkIdn~YZvFvITRqeUthp1D8QnvXPMPE2Ussr0YslpDJdvUPG4oculD648UpXUa6ucROUO~4i1mdib9vFZ22vN7juNpcN~DbpmBx6wto0Ux-AoVV71VhlMOe1LJRjZKOgGTrqD~yBvXG8NXJMgPlXHPyCtMHTSI8FiCaVX4gzUlnH-e1EhTeJSiPNmQTHgqDgxO-jecKoakHPYhrIuCN7leY242PjfGm9DZJ13fVFSR0lorurew__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PARP inhibition causes structural changes and reduces markers of oxidative stress within atherosclerotic plaques of high-fat-diet-fed ApoE−/− mice. (A) H&E stain of aortas from ApoE−/− high-fat (H-F) diet or high-fat with thieno[2,3-c]isoquinolin-5-one injections (3 mg/kg) twice a week (H-F+TIQ-A) for 12 weeks; FC, foam cells. Immunohistochemistry analysis of plaques from high-fat and high-fat with thieno[2,3-c]isoquinolin-5-one animal groups with antibodies to SMA (B), nitro-tyrosine (C), or to 8-oxo-dG (D); the rightmost panels are quantifications of immunoreactivity using Image-Pro Plus software and expressed as fold change from plaques of high-fat-diet-fed ApoE−/− mice. *P < 0.01). Bars: 3 µm.
3.2 PARP inhibition plays a protective, a neutral, or a sensitizing role against vascular cell death in response to varying inflammatory agents
Given the limitations of the mouse model of atherosclerosis in examining the different cellular responses to the inflammatory agents present with plaques through which PARP inhibition promotes its anti-atherogenic effects, we elected to utilize an ex vivo system that mimics plaque dynamics. H2O2 induced concentration-dependent cell death in wild-type HUVEC, SMCs, and FC (Figure 2A). As expected, PARP inhibition achieved pharmacologically in HUVEC or by gene knockout in FC and SMCs significantly protected against H2O2-induced death (Figure 2A). The cell-killing effects of TNF, in combination with cyclohexamide (CHX), was markedly inhibited by PARP-1 gene deletion in SMCs, however, PARP-1 gene deletion conferred no protection in FC (Figure 2B). PARP inhibition in HUVEC cells resulted in only marginal inhibition of TNF-induced cell death (Figure 2B). PARP inhibition exerted very contradictory effects in response to 7-KC treatment in the tested cell types (Figure 2C). PARP-1 gene deletion protected SMCs at all tested concentrations. In fact, PARP-1 gene deletion resulted in almost complete protection against 50 µg/mL 7-KC, the highest concentration used. PARP inhibition by TIQ-A conferred no protection to HUVEC cells against 7-KC-induced cell death even at the lowest concentration of the oxidized lipid. In sharp contrast, PARP-1 gene deletion not only failed to protect FC against 7-KC-induced death, but induced a significant sensitization of FC to the lipid as compared with their wild-type counterpart.
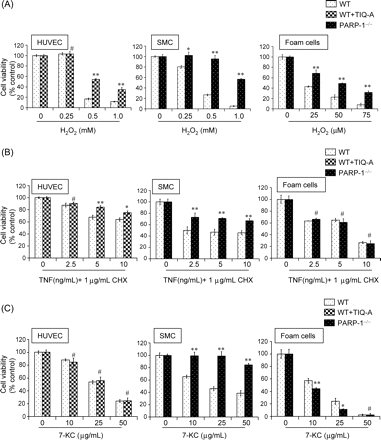
Differential responses of HUVEC, SMCs, and ex vivo-generated foam cells to increasing concentrations of H2O2 (A), TNF (B), or 7-KC (C). Cells were serum-starved for 2 h then treated with the indicated doses of the different agents for 12 h. Cell viability was measured by calcein-AM staining and is expressed as a percentage of the control value, and data are mean ± SD of triplicate analysis from a representative experiment; *P < 0.01; **P < 0.001; # non-significant (NS).
3.3 Type of inflammatory agent and type of vascular cell determine the mode of induced cell death (apoptosis or necrosis) in response to PARP inhibition: classical and novel roles for PARP-1 in mediating cell death
We next examined the nature and extent of death induced by the different inflammatory agents described above, and the effect(s) of PARP inhibition on cell death processes using annexin V staining as a marker for apoptosis and propidium iodide staining as a marker for necrosis followed by FACS analysis. PARP inhibition conferred protection to HUVEC by blocking both apoptotic and necrotic cell death in response to H2O2 (Figure 3A–C). H2O2 treatment of SMCs resulted mostly in apoptosis, which was almost completely blocked by PARP-1 gene knockout. While H2O2 induced mostly necrosis in treated FC, PARP-1 gene deletion not only protected against such mode of cell death but also altered the death process from necrosis to apoptosis. Such conversion in the mode of cell death appeared to be independent of caspase-3-like activation (data not shown). Altogether, these results clearly illustrate the differential responses of vascular cells to H2O2 exposure and suggest that PARP inhibition can block both apoptosis and necrosis. In addition, these results further expose the complexity of vascular cell death in response to H2O2.
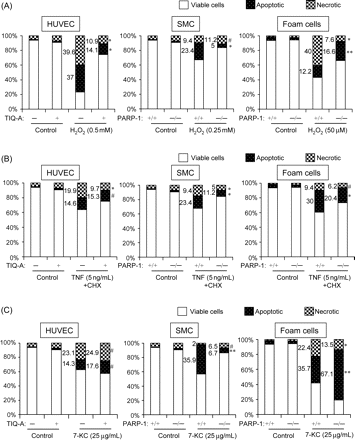
Differential induction of apoptosis and/or necrosis in HUVEC, SMCs, and ex vivo-generated foam cells treated with H2O2 (A), TNF (B), or 7-KC (C). Cells were treated as in Figure 2 after which they were subjected to staining with Annexin-V (for apoptotic death) and propidium iodide (for necrotic death) and analysed by FACS. Note the protection of PARP inhibition against H2O2-induced apoptosis and necrosis in all three cell types. In addition to protection against necrosis in FC, the mode of death was switched to apoptosis (bottom panel in A). Also, note the sensitization of PARP inhibition to 7-KC-induced apoptosis in FC (C, bottom panel). Apoptosis, necrosis, or viable cells are expressed as a percentage of the control (untreated) value, and data are mean ± SD of triplicate analysis from a representative experiment. Values (by bars) are shown when significant differences are observed; *P < 0.01; **P < 0.001; # non-significant (NS).
In response to TNF and CHX treatment, HUVEC displayed equal levels of apoptosis and necrosis and PARP inhibition treatment exerted only a marginal protective effect. PARP inhibition by gene deletion exerted similar protective effects on SMCs and FC in response to TNF and CHX treatment. The slight protection exerted by PARP-1 gene deletion on FC (Figure 3A) may be because of the high sensitivity of FACS analysis as compared with cell viability (Figure 2B).
Treatment of HUVEC with 7-KC exclusively induced necrosis, which was, surprisingly, unaffected by PARP inhibition. These results indicate that PARP inhibition does not prevent all forms of necrotic cell death and that protection from cell death may require activation of specific pathways via PARP catalytic activity. Interestingly, 7-KC induced a predominantly apoptotic response in SMCs, and this death was effectively blocked by PARP-1 gene deletion. In FC, 7-KC mainly induced apoptosis, which was exacerbated by PARP-1 gene deletion. These results are consistent with those shown with cell viability (Figure 2C) and demonstrate that PARP inhibition causes increased sensitivity of FC to 7-KC-induced toxicity. These results further illustrate the complexity of vascular cell death in that PARP inhibition may be a protective, a neutral, or a sensitizing factor.
3.4 PARP inhibition increases free cholesterol pools in FCs following 7-KC exposure: a potential cause for FC hypersensitivity?
Increased sensitivity of FC by PARP inhibition in response to 7-KC seems to be independent of ac-LDL uptake (Figure 4A). To test whether PARP inhibition interfered with the processing of cholesterol, total and free cholesterol was assessed in wild-type or PARP-1−/− FC that were treated with increasing concentrations of 7-KC. Although both groups accumulated cholesterol, only a marginal increase was observed in the total cholesterol in PARP-1−/− FC as compared with wild-type FC; however, free cholesterol content was dramatically increased in PARP-1−/− FC (Figure 4B) with concomitant decrease in esterified cholesterol. These results strongly suggest that the PARP inhibition-associated sensitivity of FC to 7-KC is because of a decrease in the detoxification (i.e. esterification) of free cholesterol.
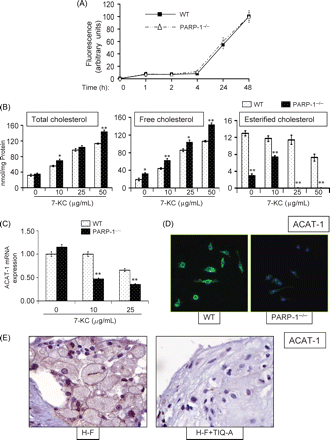
Sensitization of foam cells to 7-KC by PARP-1 gene deletion is associated with an alteration in lipid metabolism as a result of a modulation of ACAT-1 expression. (A) Lipid uptake as measured by fluorescently labeled ac-LDL is unaffected by PARP-1 gene deletion in foam cells. (B) Wild type and PARP-1−/− foam cells were treated with increasing doses of 7-KC after which intracellular concentrations of total, free, and esterified cholesterol were assessed. *P < 0.01; ** P < 0.001; # non-significant (NS). (C) Wild-type and PARP-1−/− foam cells were treated with 7-KC and total RNA was isolated, subjected to cDNA generation then to real-time PCR with primers specific to mouse ACAT-1, or β-actin; amplified PCR products were normalized and expressed as fold increase over samples derived from untreated WT cells. (D) Immunofluorescence analysis with antibodies to murine ACAT-1 and to DAPI staining (overlay) of WT or PARP-1−/− foam cells that were treated with 10 µg/mL 7-KC for 8 h; pictures were taken at the same excitation intensity. (E) Immunohistochemistry analysis of plaques from H-F and H-F+TIQ-A animal groups with antibodies to the ACAT-1.
3.5 PARP inhibition-dependent increases in free cholesterol pools during 7-KC exposure in FCs is associated with a reduction in ACAT-1 expression
ACAT-1 is the primary enzyme that is required for the conversion of toxic-free cholesterol into less toxic and soluble-esterified cholesterol in macrophages and FC.1,11 Thus, it became conceivable that PARP inhibition may negatively affect ACAT-1 expression. Indeed, ACAT-1 gene expression was severely reduced in PARP-1−/− FC after treatment with 10 µg/mL 7-KC as assessed by real-time PCR (Figure 4C). Such difference was confirmed by immunofluorescence with antibodies to mouse ACAT-1 (Figure 4D). These results were corroborated in vivo in that TIQ-A administration was found to be markedly reduced in plaques of high-fat-diet-fed ApoE−/− mice as assessed by immunohistochemistry with antibodies to ACAT-1 (Figure 4E). Together, these results suggest that the decrease in free cholesterol esterification in FC may be directly linked to a reduction in ACAT-1 expression.
3.6 PARP participation in the cell death process occurs in the presence or absence of PARP enzymatic activity (NAD utilization): novel vs. classical role of PARP-1 in cell death
The role of PARP in the process of cell death has consistently been associated with its enzymatic activity through the utilization and consequent depletion of NAD. Therefore, we examined NAD utilization in HUVEC, SMCs, and FC after a 30 min exposure to H2O2, TNF, or 7-KC. As expected, exposure to H2O2 resulted in a severe depletion of intracellular NAD in all types of vascular cells, and this depletion was markedly restored with PARP inhibition in these cells (Figure 5A). In response to TNF or 7-KC treatment, cell death in SMC, HUVEC or FC and differential effects exerted by PARP inhibition appeared to involve mechanism(s) independent of the enzymatic activity of PARP and consequent utilization of NAD. The lack of PARP activation in the latter conditions was confirmed by immunofluorescence using antibodies to the poly(ADP-ribose) moieties of modified proteins (Figure 5B).
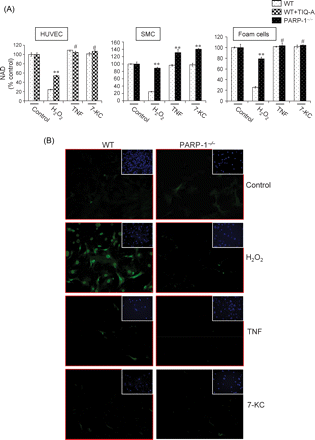
Treatment with H2O2, but not TNF or 7-KC, induces lowering of intracellular levels of NAD in vascular cells and poly(ADP-ribose) synthesis in SMCs. (A) Cells were exposed to H2O2, TNF, or 7-KC for 30 min and intracellular NAD levels were measured; the doses of the different agents are the same as those mentioned in Figure 3. Note that while H2O2 treatment induced a significant lowering of NAD, treatment with TNF or 7-KC did not cause any detectable lowering of the levels of this molecule. Values are expressed as percentage of control (untreated). *P < 0.01; ** P < 0.001; # non-significant (NS). (B) SMCs were treated as in (A) after which, they were subjected to immunofluorescence analysis with monoclonal antibodies to poly(ADP-ribose); cells were also stained with DAPI (inserts).
3.7 Protective effect of PARP-1 gene deletion against TNF- or 7-KC-induced death of SMCs involves a reduction in caspase-3 activation, an activation of the pro-survival ERK1/2 pathway and a modulation of the pro-death JNK pathway
We next examined whether PARP-1 inhibition-promoted protection against cell death was associated with blocking the activation of key factors involved in apoptosis, such as activation of caspase-3.12Figure 6A shows that, indeed, PARP-1 gene deletion significantly reduced caspase-3-like activation in SMCs treated with TNF or 7-KC. To determine such effects on caspase-3 activation in vivo using the animal model of atherosclerosis, immunohistochemical examination of the plaques suggests that PARP inhibition markedly reduced caspase-3 activation (Figure 6B) as evidenced by the significant decrease in anti-caspase-3 immunoreactivity. Overall, these in vivo results are consistent with the data attained using the cell culture system.
The lack of a clear association between PARP activation (NAD utilization) and death of SMCs in response to TNF or 7-KC led us to surmise that PARP inhibition may trigger cell survival signal transduction or prevent cell death inducing pathways to allow SMCs to resist cytotoxicity of the two inflammatory agents. Figure 6C shows that although the two agents induced a robust JNK pathway as manifested by JNK1/2 phosphorylation shortly after treatment in wild type SMCs, such activation was rather weak in TNF or 7-KC-treated PARP-1−/− SMCs. Further, a robust and transient activation of the ERK pathway as manifested by ERK1/2 phosphorylation was observed in response to TNF or 7-KC exposure of PARP-1−/− SMC (Figure 6D). Little to no ERK1/2 phosphorylation was detected in wild-type SMC under these conditions. These results suggest that PARP inhibition may exert protection against cell death independently from NAD utilization but through the prevention of a critical signal transduction pathway that participates in cell death as well as through the induction of a critical signal transduction pathway that participates in cell survival.
4. Discussion
In the present study, we paired the same animal model with a cell culture model mimicking plaque dynamics to provide detailed evidence for the intricate mechanisms by which PARP inhibition promotes atherosclerotic plaque stability. PARP inhibition reduced traits of plaque disruption by decreasing oxidative DNA damage, iNOS-associated protein nitration. In addition, inhibition of PARP resulted in atrophied features in FC in the animal model. PARP-1 gene deletion protected vascular cells from both necrosis and apoptosis induced by H2O2 or TNF. When necrosis was the major mode of cell death, PARP-1 gene deletion not only conferred protection to the treated cells but altered the mode of cell death from necrosis to apoptosis. Similarly, PARP-1 gene deletion protected SMCs against 7-KC. In sharp contrast, PARP inhibition by either TIQ-A treatment or gene deletion did not protect FCs from 7-KC and actually caused a significant sensitization of these cells to this agent. The increased sensitivity of PARP-1−/− FC to 7-KC was associated with an alteration in cholesterol metabolism with an increase of toxic-free cholesterol with a concomitant decrease in esterified cholesterol. This defect in cholesterol metabolism appeared to result from the defective expression of the macrophage-specific factor ACAT-1. These results not only present PARP inhibition as a potential strategy for atherosclerotic plaque stabilization but also challenge the current understanding of the role of PARP in the process of cell death. Taken together, these results indicate that PARP inhibition decreases the inflammatory response and selectively promotes FC death while protecting endothelial and SMCs from injury by oxidants or cytokines. These features of PARP inhibition are highly desirable for a therapeutic strategy to stabilize or reduce atherosclerosis plaques.13
The role of PARP in cell death has been exclusively associated with necrosis4 and viewed as dispensable for apoptosis. Earlier investigations in our laboratory led us to propose that PARP is involved in apoptotic cell death in some situations including in response to TNF.14 Interestingly, PARP-1 gene deletion reduced caspase-3-like activation in FC after treatment with 7-KC (data not shown), which may correlate with the observed dying or dead cells (see arrows in Figure 5B) in the vicinity of necrotic cores of plaques from the TIQ-A-treated mice suggesting a potential caspase-3-independent death process. Interestingly, the role of PARP in cell death and the protective effects associated with its inhibition can be independent of the enzymatic activity associated with NAD utilization. This PARP involvement was associated with a stimulation of alternative pathways through the activation of JNK. The protective effect of PARP inhibition was associated with a stimulation of the ERK1/2 pathway. The observation that PARP inhibition promotes death of FC and protects against death of SMCs and endothelial cells is clinically relevant as these traits are highly desirable for plaque stability and regression. It is important to note that some of the results of the current study were attained using an in vitro system whose limitations restrict extrapolations to what actually occurs in atherosclerotic plaques of our animal model or in human plaques (Figure 6) .
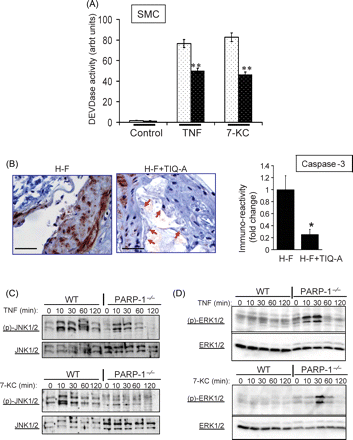
PARP-1 gene deletion reduces caspase-3-like activity, reduces JNK phosphorylation, and induces ERK1/2 phosphorylation in response to TNF or 7-KC in SMCs. (A) Cells were treated with TNF (and CHX) or 7-KC for 6 h after which protein extracts were assayed for caspase-3-like (DEVDase) activity. **P < 0.001. (B) Immunohistochemistry analysis of plaques from H-F and H-F+TIQ-A animal groups with antibodies to the active peptide of caspase-3; the rightmost panel is a quantification of immunoreactivity using Image-Pro Plus software and expressed as fold change from plaques of H-F diet-fed ApoE−/− mice. * difference from H-F diet-fed ApoE−/− mice, P < 0.01. Bars: 3 µm. Cells were treated as in (A) for the indicated time intervals after which protein extracts were subjected to Western immunoblot analysis with antibodies to pJNK1/2 or JNK1/2 (C) or pERK1/2 or ERK1/2 (D).
The mechanism by which PARP inhibition promotes death of FC is rather intriguing. ACAT-1 is thought to be responsible for FC formation and the subsequent progression of atherosclerosis.1,11,15 In macrophages, ACAT-1 is responsible for lowering the cytotoxicity of free cholesterol by conversion to cholesterol esters. The decrease in ACAT-1 expression following PARP inhibition may be directly related to the increase in free cholesterol detected in PARP-1−/− FC (Figure 4) and TIQ-A-treated wild-type cells (data not shown) and the subsequent sensitization to death. ACAT-1 inhibition in FC results in accumulation of free cholesterol, which in turn, induces cell death by apoptosis.16 It is noteworthy that PARP-1 gene deletion did not sensitize SMCs to 7-KC exposure, which may be directly related to the inability of these cells to express ACAT-1.17
Overall, these results of this study indicate that PARP inhibition affects atherosclerotic plaque dynamics and that these alterations are highly desirable for the control of plaque progression and promotion of stability. Thus, manipulation of PARP in patients may offer a mechanism to control atherosclerotic plaques. Since PARP inhibition selectively protects SMCs and endothelial cells from death, promotes cell death in FC, and inhibits pro-inflammatory factors such as MCP-1 and ICAM-1, this enzyme is a promising therapeutic target with many important modes of action.
Funding
National Institutes of Health (HL072889 and 1P20RR18766) to HB.
Acknowledgements
We thank Drs. E. Songu-Mize, B. Worthylake, and L. Harrison-Bernard for their help with cell culture, fluorescence microscopy, and quantification of immunoreactivity, respectively.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}