





Abstract
Objective: Abdominal aortic aneurysms (AAAs) are characterized by chronic inflammation which contributes to the remodeling and eventual weakening of the vessel wall. Increased cyclooxygenase-2 (COX-2) expression is detected in human aneurysmal tissue and is suggested to contribute to the disease. The aim of the current study was to define the role of COX-2 expression in the development of AAAs, using a model of the disease.
Methods: AAAs were induced in mice by chronic angiotensin II infusion, and were analyzed following 3, 7, 21 or 28 days of the infusion. AAA incidence and severity, together with the expression of inflammatory markers, were compared between abdominal aortas from COX-2-deficient mice and their wild-type littermate controls.
Results: The AAA incidence in COX-2 wild-type mice was 54% (13/24), whereas AAAs were not detected in COX-2-deficient mice (0/23) following 28 days of angiotensin II infusion. The genetic deficiency of COX-2 also resulted in a 73% and 90% reduction in AAA incidence following 7 and 21 days of angiotensin II infusion, respectively. In COX-2 wild-type mice, COX-2 mRNA expression in the abdominal aorta was induced by angiotensin II beginning 3 days following initiation of the infusion, which continued throughout progression of the disease. Abundant COX-2 protein expression was detected in medial smooth muscle cells adjacent to the AAAs. The deficiency of COX-2 significantly attenuated mRNA expression in the abdominal aorta of the macrophage marker CD68, and the inflammatory cell recruitment chemokines, monocyte chemotactic protein-1 and macrophage inflammatory protein-1α.
Conclusions: Our findings suggest that increased COX-2 expression in smooth muscle cells of the abdominal aorta contributes to AAA formation in mice by enhancing inflammatory cell infiltration.
1. Introduction
Abdominal aortic aneurysms (AAAs) are an increasing health concern, particularly in the aging population. Chronic inflammation is a feature of the majority of human AAAs, and inflammatory cells are thought to contribute to the tissue destruction that leads to the weakening and eventual rupture of the vessel [1]. Although a variety of inflammatory cells are increased in aneurysmal tissue, macrophages are believed to have a pronounced role in the pathogenesis of AAAs [2–4].
Although pharmacological treatments are not currently available, the use of non-steroidal anti-inflammatory drugs (NSAIDs) has been suggested to reduce AAA development or expansion in humans and animal models [5]. A well characterized mechanism of action of all NSAIDs is the metabolic inhibition of the cyclooxygenases (COXs), which are present as two isoforms, termed COX-1 and COX-2 [6]. Both COX isoforms metabolize arachidonic acid into prostaglandin (PG)H2, which is further subjected to non-enzymatic or enzymatic conversion to form biologically active PGs and thromboxane A2, collectively known as prostanoids [6]. In human aneurysmal tissue, the expression of COX-2 and resultant PG production have been shown to be increased as compared to normal aorta, whereas COX-1 expression is not significantly altered [5,7]. Because of the predominant expression of COX-2 in this disease, inhibitors selective for this isoform have been proposed as a treatment for AAAs [5,7].
Similar to the human disease, experimental models have suggested a role for COX activity in contributing to AAA expansion. In a rat elastase perfusion model, treatment with the nonselective COX isoform inhibitor indomethacin reduced AAA growth [8,9]. Another widely used experimental model utilizes chronic infusion of angiotensin II (AngII) to induce AAAs in hyperlipidemic and nonhyperlipidemic mice [10,11]. AngII-induced AAA development is characterized by macrophage infiltration into the vessel wall and the elimination of AngII-dependent monocyte/macrophage-mediated inflammation suppresses AAA formation [12,13]. Using this model, we have recently shown that treatment with the COX-2-selective inhibitor celecoxib, but not a COX-1-selective inhibitor, significantly reduces the incidence and severity of AngII-induced AAAs in both hyperlipidemic and nonhyperlipidemic mice [14]. However, the beneficial effects of celecoxib in treating another model of cardiovascular disease have been suggested to occur through mechanisms that are independent of COX-2 inhibition [15]. Furthermore, rofecoxib, another COX-2 inhibitor which does not act on the non-COX-2 targets inhibited by celecoxib, has recently been shown not to reduce AAA development in another animal model [16]. Therefore, to eliminate potential effects produced by nonspecific pharmacological inhibition, we utilized COX-2-deficient mice to examine the role of COX-2 in AngII-induced AAA development.
2. Methods
2.1. Animals
Eight to ten-week-old COX-2-wild-type (COX-2+/+) and COX-2-deficient (COX-2−/−) mice of a hybrid C57BL/6J×129/Ola (C57/129) genetic background, as originally described by Morham et al, [17] were obtained from Taconic Farms Inc. COX-2−/− mice and COX-2+/+ matched controls were produced from crossing the COX-2 heterozygous mutant line that has been maintained for more than 35 generations. The COX-2+/+ and COX-2−/− mice used in these studies were on a nonhyperlipidemic background and were housed under barrier conditions with food and water provided ad libitum. All studies were performed with the approval of the University of Kentucky Institutional Animal Care and Use Committee and conform with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.2. AngII infusion
Mice were anesthetized (25 mg/kg ketamine and 2.5 mg/kg xylazine, single dose IP) and AngII (1000 ng/kg per min; Sigma, St. Louis, MO) or saline was administered by Alzet osmotic minipumps (model 2004) implanted subcutaneously, as described previously [10]. Mouse body weight was measured weekly throughout the time-course of the experiment.
2.3. Vascular pathology
Aneurysms in the abdominal aorta were quantified by the percent incidence, whereby the abdominal aortas with external diameters of ≥1.5 mm were considered an AAA. Similar to previous studies, [12] we observed that aortic rupture and resulting mortality occurred in approximately 10% of mice following AngII infusion, and these animals that died prematurely were not included in the AAA incidence calculation. AAA severity was determined by measuring the external diameter of the supra-renal abdominal aorta with the following classification, Type 1: 1.5–2 mm, Type 2: 2–3 mm, Type 3: greater than 3 mm, and by measuring the wet weights of the abdominal aortas.
2.4. Tissue collection and RNA isolation
Following euthanasia at the designated time-points, aortas were excised from COX-2+/+ and COX-2−/− mice and divided into arch, thoracic and abdominal segments and stored in RNALater (Sigma). Total RNA was extracted from the individual abdominal segments using RNeasy Mini Kits (Qiagen, Valencia, CA).
2.5. Quantitation of mRNA expression
Quantitative gene expression was performed in a two-step RT-PCR using Superscript II (Invitrogen) and fluorogenic 5′ nuclease chemistry (Taqman chemistry). Primer/probe assays for COX-2, microsomal PGE2 synthase (mPGES-1), CD68, monocyte chemotactic protein-1 (MCP-1) and macrophage inflammatory protein-1α (MIP-1α) were purchased from Applied Biosystems (Foster City, CA). mRNA expression of the constitutively expressed housekeeping gene hypoxanthine phospho-ribosyl transferase (HPRT) was also quantitated as an internal normalizing gene [18,19]. There was no significant difference in HPRT mRNA levels between saline- or AngII-infused mice or throughout the time-course of the study, when compared to other housekeeping genes (data not shown). A relative standard curve using cDNA from TPA-treated skin, lung tissue, or peritoneal macrophages, as appropriate, was run within the same reaction. The quantity of mRNA for the gene of interest was extrapolated from the respective standard curve and normalized to HPRT levels. Because the expression analysis for each gene of interest was performed on separate reaction plates, and because the primers for each gene of interest were of different efficiencies, comparison of expression levels between genes of interest was not performed.
2.6. Histology
Mice were perfused with a 4% paraformaldehyde/5% sucrose solution and the aortas were dissected and divided into segments, as described above. Tissues were processed (paraffin embedded) for histological analysis. Serial sections (6 μm) of the abdominal aortas were used for immunohistochemical analysis for COX-2 (primary antibody from Cayman Chemical, Ann Arbor, MI), smooth muscle α-actin (primary antibody from Dako Cytomation, Carpinteria, CA) and Mac-3 (primary antibody from BD PharMingen, San Diego, CA). Antibody binding was detected with the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) and di-amino benzidine staining (brown) using manufacturer's instructions. Macrophage infiltration within the abdominal aorta was quantitated by determining the ratio of the number of Mac-3 positive cells to the total number of hematoxylin positive nuclei per section.
2.7. Cholesterol measurement
Blood samples were obtained at time of sacrifice by cardiac puncture. Serum total cholesterol was determined using enzymatic assay kits (Wako Chemical Co, Richmond, VA) and the results are reported as the mean±SEM.
2.8. Statistical analyses
The mean and SEM were calculated for each parameter with an individual aorta from a single mouse considered as an n of 1. Significant differences in AAA incidence among different groups was determined using χ2 analysis. Statistical significance of quantitative PCR data was determined using the Mann Whitney test. Differences were considered statistically significant at P<0.05.
3. Results
3.1. Reduced AAA formation in mice genetically deficient in COX-2
To determine the role of COX-2 in AAA formation, the incidence of AngII-induced AAAs was compared between mice that express COX-2 and mice with the systemic genetic deficiency of COX-2 that we have previously developed [17]. Male mice were infused with saline or AngII and were sacrificed after 28 days for determining AAA incidence and severity. Similar to previous reports with hyperlipidemic mice, [10,12] AngII-induced aneurysms developed in the abdominal aorta, anterior to the branching of the renal arteries. AngII infusion induced a 54% (13/24) incidence of AAAs in COX-2+/+ mice following 28 days of AngII infusion (Fig. 1A). In contrast, AAAs were not observed in COX-2−/− mice (0/23) following 28 days of AngII infusion (Fig. 1A).
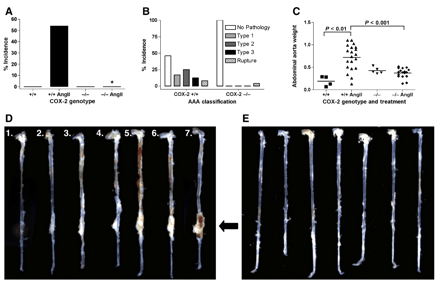
AngII-induced AAA formation in COX-2+/+ and COX-2−/− mice. (A) AAA incidence and (B) AAA severity following 28 days of AngII infusion. n≥20, *, P<0.001, χ2 test. (C) Abdominal aorta weight (normalized to total body weight (μg aorta/g body weight) for saline- (▪) or AngII-treated (▴) COX-2+/+ mice, and saline- (▾) or AngII-treated (♦) COX-2−/− mice. P values determined by Mann Whitney test. Representative aortas from AngII-infused (D) COX-2+/+ mice (1, 2: no pathology; 3, 4: Type 1; 5, 6: Type 2; 7: Type 3) and (E) COX-2−/− mice following 21 days of AngII infusion. Arrow indicates abdominal aorta.
AAA severity was categorized from Type 1 to Type 3, as determined by the external diameter of the supra-renal abdominal aorta (Fig. 1B). The abdominal aorta from saline-infused mice did not exceed 1 mm, whereas the same aortic region from AngII-infused COX-2+/+ mice ranged from ≤1 mm (no pathology), to a pronounced bulbous of ≥3 mm in diameter (Type 3). A low incidence of aortic rupture and mortality was observed in both the COX-2+/+ (2/26) and COX-2−/− (1/24) mice prior to completion of the 28 days of AngII infusion (Fig. 1B). As an additional indicator of AngII-induced vascular pathology, wet weights of the abdominal aortic segments were determined. In COX-2+/+ mice, AngII infusion significantly increased the abdominal aorta weight as compared to the abdominal aortas of saline-infused mice (Fig. 1C). In contrast, AngII infusion produced no such increase in abdominal aorta weight in COX-2−/− mice when compared to saline-infused controls (Fig. 1C). In mice that were not infused with AngII, there was no significant difference in abdominal aortic weights between COX-2+/+ and COX-2−/− mice (Fig. 1C).
We also determined the effect of COX-2 deficiency on abdominal aortic pathology following 3, 7 and 21 days of AngII infusion. Similar to the 28 day time-point, the genetic deficiency of COX-2 significantly reduced AAA incidence following 7 or 21 days of AngII infusion (7 day infusion, COX-2+/+: 43% [15/35] versus COX-2−/−: 12% [3/26], P<0.01, χ2 test; 21 day infusion, COX-2+/+: 37% [11/30] versus COX-2−/−: 4% [1/28, P<0.01, χ2 test). When analyzed following 3 days of AngII infusion, the AAA incidence was lower than the other time-points and was not significantly different between COX-2+/+ and COX-2−/− mice (COX-2+/+: 31% [5/16] versus COX-2−/−: 6% [1/16], P=0.07, χ2 test). Abdominal aorta pathology following 21 days of AngII infusion was similar to the 28 day results, with AAAs developing with a range of severities in COX-2 wild-type mice (Fig. 1D) and a significant reduction in AAA formation in mice deficient in COX-2 (Fig. 1E). These findings demonstrate that the genetic deficiency of COX-2 dramatically reduces susceptibility to AngII-induced AAA formation.
3.2. Increased expression of COX-2 during AngII-induced AAA formation
To identify the stage of AAA formation and/or progression when COX-2 may contribute to the disease, we examined the temporal pattern of COX-2 mRNA expression throughout the progression of AngII-induced AAAs. COX-2+/+ mice were infused with saline or AngII and sacrificed at 3, 7 or 21 days for analysis of COX-2 mRNA expression in the abdominal aorta by real-time PCR. AngII infusion significantly increased the levels of COX-2 mRNA at experimental days 3, 7 and 21, as compared to saline infusion (Fig. 2A–C). These findings indicate that COX-2 mRNA expression is induced at an early stage of AAA formation and the induction continues through progression of the disease.
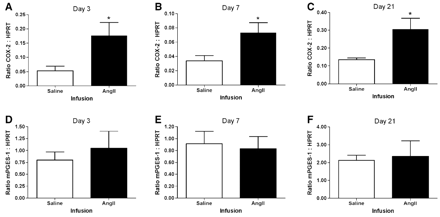
COX-2 and mPGES-1 mRNA expression in the abdominal aorta of COX-2+/+ mice. COX-2 mRNA expression of saline-(clear bars) and AngII-(filled bars) infused mice at (A) day 3, (B) day 7 and (C) day 21 post-infusion. mPGES-1 mRNA expression of saline-(clear bars) and AngII-(filled bars) infused mice at (D) day 3, (E) day 7 and (F) day 21 post-infusion. COX-2 or mPGES-1 mRNA expression was normalized to HPRT levels. Data represent mean±SEM. n≥6, *, P<0.05, Mann Whitney test.
Following the induction of COX-2 expression, the formation of PGs is increased by specific downstream prostanoid synthases [20]. Of the prostanoid synthases known to synthesize PGE2, only mPGES-1 shows inducible expression [20]. Therefore, we quantitated mPGES-1 mRNA at the same time-points when COX-2 mRNA was significantly increased by AngII infusion. In contrast to COX-2 expression, mPGES-1 mRNA expression was not induced on days 3, 7 or 21 following AngII infusion, when compared to the saline-treated controls (Fig. 2D–F). These findings suggest that increased expression of mPGES-1 may not contribute to AngII-induced AAA formation.
To determine cellular localization of COX-2, we examined expression by immunohistochemistry in the abdominal aorta following AngII infusion of mice for 21 days. Significant COX-2 expression was detected in the medial layer adjacent to AAAs, while more diffuse expression was observed in AAAs from wild-type mice (Fig. 3A). Higher magnification shows COX-2 expression concentrated within individual nuclei of medial SMCs (Fig. 3B). COX-2 expression was not observed in abdominal aorta from COX-2−/− mice (Fig. 3C). The region of the medial layer positive for COX-2 expression also expressed the smooth muscle cell (SMC) marker α-actin (Fig. 3D). The macrophage marker protein Mac-3 was detected in macrophages within the adventitia, but not in the COX-2-positive region of the medial layer (Fig. 3E). Therefore, SMCs of the abdominal aorta may be the cell type primarily responsible for the expression of COX-2 which contributes to AAA formation.
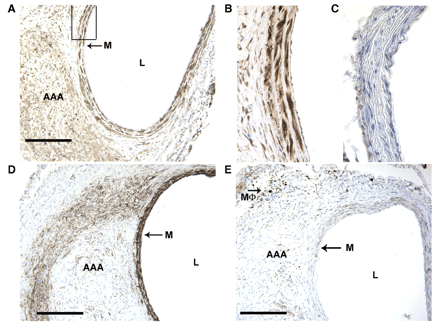
Immunohistochemical analysis of COX-2 expressing cell types in AAAs. COX-2 expression in abdominal aorta from COX-2+/+ at (A) ×100 magnification with boxed region shown in (B) at ×300 magnification and (C) COX-2−/− mice. (D) Smooth muscle α-actin expression at ×100 magnification. (E) Macrophage marker (Mac-3) expression at ×100 magnification. Brown staining indicates COX-2, actin, or Mac-3 expression and sections are counterstained with hematoxylin (blue). Scale bar 200 μm. Lumen (L), media (M), abdominal aortic aneurysm (AAA), and macrophage (Mϕ).
3.3. COX-2 deficiency attenuates AngII-induced macrophage infiltration into the abdominal aorta
Macrophage infiltration into the wall of the abdominal aorta is an early event following AngII infusion [12]. Therefore, we examined macrophage infiltration as a potential mechanism by which COX-2 contributes to AAA formation. CD68 mRNA was used as a quantitative marker of macrophage accumulation in the vessel wall. In the abdominal aorta of AngII-infused mice, CD68 mRNA expression was significantly increased on experimental days 3, 7 and 21, as compared to mice infused with saline (Fig. 4A–C). CD68 mRNA expression was significantly attenuated in COX-2−/− mice, as compared to COX-2+/+ mice, on experimental days 3, 7 and 21 following the initiation of AngII infusion (Fig. 4D–F). Immunohistochemical analysis of the abdominal aorta from AngII-infused COX-2+/+ mice, identified numerous Mac-3-positive cells within the adventitia, and fewer Mac-3-positive cells within the medial layer (Fig. 4G,H). As compared to the abdominal aorta of COX-2+/+ mice, the number of Mac-3 positive cells was significantly reduced in AngII-infused COX-2−/− mice (Fig. 4 I and I inset).
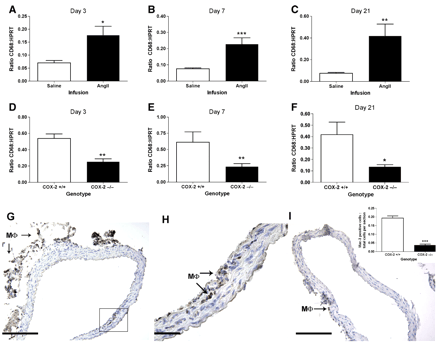
CD68 mRNA and Mac-3 protein expression in the abdominal aorta. Saline-(clear bars) and AngII-(filled bars) infused COX-2+/+ mice at (A) day 3, (B) day 7 and (C) day 21 post-infusion. AngII-infused COX-2+/+(clear bars) and COX-2−/− (filled bar) mice at (D) day 3, (E) day 7 and (F) day 21 post-infusion. Data represent mean±SEM. n≥6, *, P<0.05, **, P<0.01, ***, P<0.001, Mann Whitney test. Immunohistochemical analysis of Mac-3 expression in the abdominal aorta following 7 day AngII infusion of (G) COX-2+/+ mice (×100, scale bar 200 μm), (H) COX-2+/+ mice (×300, scale bar 50 μm) of boxed area in G, (I) COX-2−/− mice (×100). (I inset) Quantitation of Mac-3 positive cells per total number of cells in each section for COX-2+/+ and COX-2−/− mice. Data represent mean±SEM. n=5, ***, P<0.001, unpaired 2-tailed Student's t test. Brown staining indicates Mac-3 expression and sections are counterstained with hematoxylin (blue). Arrows indicate Mac-3 positive cells (Mϕ).
To identify a mechanism by which COX-2 may contribute to macrophage infiltration, we examined the effect of COX-2 deficiency on the mRNA expression of chemokines during the initial stage of AAA formation. The deficiency of COX-2 significantly attenuated the expression of MCP-1 and MIP-1α mRNA in the abdominal aorta at both the 3 and 7 day time-points following the initiation of AngII infusion (Fig. 5A–D). These data suggest that attenuated chemokine expression resulting in reduced macrophage accumulation contributes to the diminished AAA formation that we observed in COX-2-deficient mice.
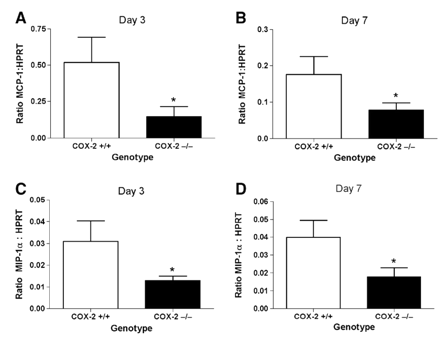
MCP-1 and MIP-1α mRNA expression in the abdominal aorta. MCP-1 mRNA expression in AngII-infused COX-2+/+(clear bars) and COX-2−/− (filled bar) mice at (A) day 3, (B) day 7 post-infusion. MIP-1α mRNA expression in AngII-infused COX-2+/+(clear bars) and COX-2−/− (filled bar) mice at (C) day 3, (D) day 7 post-infusion. Data represent mean±SEM. n≥6, *, P<0.05, Mann Whitney test.
3.4. COX-2 deficiency does not reduce plasma cholesterol concentrations
In the C57BL/6 strain of mice, the incidence of AngII-induced AAAs is increased by hyperlipidemia [11,21]. Therefore, we examined if decreased serum cholesterol levels in COX-2−/− mice accounted for the observed reduction in AAA incidence. However, there was no significant difference in the total serum cholesterol concentrations between COX-2+/+ and COX-2−/− mice during the course of the AngII infusion (Table 1).
Total serum cholesterol in AngII-infused COX-2+/+ and COX-2−/− mice
| AngII infusion (days) | Total serum cholesterol (mg/dl) | |
|---|---|---|
| COX-2+/+ mice | COX-2−/− mice | |
| 3 | 128±16 | 171±18 |
| 7 | 118±8 | 167±27 |
| 21 | 136±8 | 171±19 |
| 28 | 146±11 | 190±19 |
| AngII infusion (days) | Total serum cholesterol (mg/dl) | |
|---|---|---|
| COX-2+/+ mice | COX-2−/− mice | |
| 3 | 128±16 | 171±18 |
| 7 | 118±8 | 167±27 |
| 21 | 136±8 | 171±19 |
| 28 | 146±11 | 190±19 |
Data represent mean±SEM, n≥9.
Total serum cholesterol in AngII-infused COX-2+/+ and COX-2−/− mice
| AngII infusion (days) | Total serum cholesterol (mg/dl) | |
|---|---|---|
| COX-2+/+ mice | COX-2−/− mice | |
| 3 | 128±16 | 171±18 |
| 7 | 118±8 | 167±27 |
| 21 | 136±8 | 171±19 |
| 28 | 146±11 | 190±19 |
| AngII infusion (days) | Total serum cholesterol (mg/dl) | |
|---|---|---|
| COX-2+/+ mice | COX-2−/− mice | |
| 3 | 128±16 | 171±18 |
| 7 | 118±8 | 167±27 |
| 21 | 136±8 | 171±19 |
| 28 | 146±11 | 190±19 |
Data represent mean±SEM, n≥9.
4. Discussion
In the current study, we examined the role of COX-2 expression in AAA formation, using an animal model of the disease induced by AngII. Following 28 days of AngII infusion, a significant incidence of AAAs of varying severity was observed in COX-2+/+ mice (54%), whereas AAAs were not observed (0/23) in COX-2−/− mice. There was also a significant reduction in AAA incidence in COX-2−/− mice, as compared to COX-2+/+ mice, following 7 or 21 days of AngII infusion. A low incidence of AAAs was also detected earlier in the time-course of AngII infusion at the 3 day time-point, although the difference between COX-2+/+ and COX-2−/− mice was not statistically significant. The AAAs which formed in the COX-2−/− mice were all of the least severe classification (Type 1). The observation that COX-2-deficient mice showed reduced AAA incidence and severity at multiple stages of the disease, suggests that COX-2 expression contributes to the initial formation and the early progression of AAAs.
Previous reports with the AngII model of AAAs have shown that approximately 10% of the mice die from an aortic rupture during the first 10 days of the infusion [12]. In the current study, we also observed a low incidence of aortic rupture early in the course of the AngII infusion. However, unlike the development of AAAs, the incidence of rupture and resulting mortality during the first 10 days of the AngII infusion was not significantly different between COX-2+/+ and COX-2−/− mice. Thus, although COX-2 is critical to the pathological remodeling associated with AAA formation or expansion, the aortic rupture that occurs in this model may be independent of COX-2 expression.
In aortic SMCs, AngII is known to be a potent inducer of COX-2 transcription [22,23]. We previously reported that in hyperlipidemic mice, AngII infusion induces significant COX-2 expression that is primarily localized in SMCs of the abdominal aorta [14]. In the current study, we show that in nonhyperlipidemic mice COX-2 expression was concentrated in SMCs of the medial layer and more diffuse expression was detected within the AAAs and in macrophages (Mac-3 positive) of the adventitia. Furthermore, AngII increased COX-2 mRNA expression as early as 3 days following initiation of the infusion. These studies suggest that the AngII-dependent induction of COX-2 within SMCs of the abdominal aorta is an early event leading to the pathological remodeling that contributes to AAA formation.
Inflammatory cell infiltration into the vessel wall of the abdominal aorta is the first discernable pathology associated with AngII-induced AAA development [12]. Previous studies using another model of cardiovascular disease, have shown that prostanoids derived from the activity of COX-2 contribute to vascular inflammation [24]. The current report shows that the deficiency of COX-2 significantly reduces macrophage infiltration into the abdominal aorta following AngII infusion. Macrophage recruitment is stimulated by chemokines such as MCP-1 that are produced by the endothelium and SMCs of the vessel, and MCP-1 expression has been shown to be increased in the abdominal aorta during AngII-induced aneurysm formation [25,26]. In addition, expression of the monocyte chemokine MIP-1α has been shown to be increased during leukotriene-dependent AAA formation in mice [27]. We observed that the deficiency of COX-2 significantly reduced both MCP-1 and MIP-1α mRNA expression during early AAA development. Previously, it has been reported that leukocyte-specific deficiency of CCR2, the receptor for MCP-1, significantly reduces AngII-induced macrophage infiltration and AAA formation, without reducing MCP-1 expression in the aorta [13]. Another report utilizing the AngII-induced AAA model in mice has recently shown that AngII increases mRNA expression of MCP-1 and CCR2 in aorta, prior to increased macrophage content within the vessel wall [26]. These findings indicate that macrophages do not contribute to MCP-1 production in this model and suggest that another cell type, potentially SMCs, is the primary source of this chemokine during AngII-induced AAA formation. Therefore, the induction of COX-2 that we observed during AAA development may initiate macrophage infiltration into the abdominal aorta by increasing SMC-dependent production of recruitment chemokines.
Although hypercholesterolemia has been described as a risk factor for development of AAAs in humans, other factors such as smoking, male gender and advanced age are more significant [28,29]. A recent AAA screening study also suggests that elevated plasma lipid levels and atherosclerotic burden are not associated with expansion of small AAAs [30]. In the AngII-induced model, the AAA incidence in the C57BL/6J mouse strain is increased when crossed into a hyperlipidemic background [10,11,31]. However, we recently reported that AngII infusion induces a similar incidence of AAAs in hyperlipidemic Apoe−/− mice as that produced in the nonhyperlipidemic wild-type mice used in the current study [14]. Additionally, our previous report showed that celecoxib treatment was equally effective at reducing AAA formation in both the hyperlipidemic and nonhyperlipidemic mouse strains [14]. In the current studies, there was no significant difference in the total serum cholesterol levels between COX-2+/+ and COX-2−/− mice following 3, 7, 21 or 28 days of AngII infusion. Therefore, the effectiveness of the COX-2 deficiency in protecting against AngII-induced AAA formation was not the result of an indirect effect of the COX-2 deficiency on reducing serum cholesterol levels.
Studies examining changes in systolic blood pressure with the AngII-induced AAA model have reported either no measurable increase or a modest increase during the AngII infusion. Significant AngII-induced AAA development is known to occur without a measurable increase in blood pressure [10]. Another recent report utilizing a range of AngII doses and a hypertensive agent unrelated to AngII, suggests that increased systolic blood pressure is not a mechanism contributing to AngII-induced AAA formation [26]. We previously reported that the effectiveness of COX-2-selective inhibitor treatment on reducing AngII-induced AAA formation was not associated with a reduction in blood pressure [14]. Furthermore, the genetic deficiency of COX-2 does not result in any alterations of systolic blood pressure in untreated mice, [32] and COX-2 inactivation enhances the acute pressor response of AngII [33]. Therefore, based these previous observations, we do not expect that the attenuation of AAA pathology in COX-2-deficient mice resulted from a reduction in blood pressure.
Numerous stimuli acting on different cell types of the vessel wall may contribute to the inflammatory response that leads to AAA development. Recently, the intracellular signaling pathway involving c-Jun N-terminal kinase (JNK) has been shown to be activated in human AAA tissue and is essential in animal models of AAA development, including AngII-induced AAAs in mice [34]. Another signaling pathway involving the serine/threonine kinase Akt, has been shown to be necessary for AngII-dependent pathology following injury of the vessel wall [35]. Surprisingly, both JNK and Akt have recently been identified as targets of the COX-2-selective inhibitor celecoxib. Furthermore, the inhibition of JNK and Akt phosphorylation by celecoxib occurs through a mechanism independent of COX-2 inhibition [15,36]. Collectively, these studies suggest there is the potential for alternative mechanisms of the beneficial effects of celecoxib in the reduction of AAAs that we recently reported [14]. However, by comparing AAA formation in mice genetically deficient in COX-2 with their littermate wild-type controls, the current study has unequivocally determined that COX-2 expression significantly contributes to AngII-induced AAA development in mice.
Previous reports have shown that increased COX-2 expression and PGE2 production occur in aneurysmal tissues [5,7]. Our recent report also showed that AngII infusion significantly increases the ex vivo synthesis of PGE2 in the abdominal aortic segment [14]. The substrate PGH2 required for the formation of PGE2 is synthesized by both COX-1 and COX-2 [6]. The formation of PGE2 from PGH2 may occur either non-enzymatically, [37] or enzymatically by the activity specific terminal prostanoid synthases [20]. Of the prostanoid synthases known to synthesize PGE2, only mPGES-1 shows inducible expression [20]. However, there are no reports of increased mPGES-1 expression during AAA formation, and unlike the increase in COX-2, we observed no increase in mPGES-1 mRNA expression during AngII-induced AAA formation in the current studies. Therefore, increased production of PGE2 contributing to COX-2-dependent AAA formation may be due to the activity of constitutively expressed mPGES-1 or other PGE2 synthases, or due to non-enzymatic formation.
Although the current study identifies an important role for COX-2 expression in a model of AAA formation, serious questions have been raised about the safety of medications which selectively inhibit this isoform [38,39]. With the recent determination of increased adverse cardiovascular events in patients following long-term treatment with COX-2-selective inhibitors, there is considerable interest in identifying targets downstream of COX-2. Individual prostanoid receptors have been proposed as alternative targets for blocking pathological effects of increased COX-2 activity [40]. Human aneurysmal tissue has been shown to express a variety of prostanoid receptors, and activation of the PGE2 receptor EP4 has been proposed to increase production of inflammatory mediators contributing to AAA pathology [41]. Further studies are needed to examine the effects of inactivating individual prostanoid receptors involved in COX-2-dependent AAA formation. Antagonists which target the specific prostanoid receptor(s) contributing to AAAs may lack the adverse cardiovascular effects of COX-2-selective inhibition, and thereby provide a safe and effective treatment.
References
Author notes
JMG and DBT contributed equally to this work.
Time for primary review 30 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}