





Abstract
Objective: CD36 is a receptor, whose expression increases during the differentiation of monocytes to macrophages, playing a key role in the phagocytosis of apoptotic cells and in the formation of foam cells during atherosclerosis. Recently, it has been described that ligands of PPARγ induce CD36 expression and inhibit cyclooxygenase expression in macrophages. Our aim was to study whether the reduction of endogenous prostaglandin production could modify CD36 expression in macrophages and to outline the potential mechanism.
Methods and results: CD36 expression was measured by flow cytometry in THP-1 cells differentiated to macrophages that had been incubated with aspirin (ASA) alone or in combination with PGE2, sulprostone (EP1/EP3 agonist), butaprost (EP2 agonist,) and PGE1 alcohol (EP2/EP4 agonist). Aspirin induced CD36 expression. Only PGE2 and PGE1 alcohol completely abolished CD36 induction by aspirin, whereas butaprost strongly reduced it. BADGE (a PPARγ antagonist) or diclofenac (a PPARγ antagonist and a cyclooxygenase inhibitor) in aspirin-incubated cells did not reduce CD36 induction. On the other hand, aspirin also induced the expression of SR-BI and ABCA1, an HDL receptor and an HDL formation-related protein, respectively.
Conclusions: Aspirin produces an increase of CD36 expression in THP-1 macrophages by a PGE2-dependent mechanism. The PGE2 receptors implicated in CD36 modulation by ASA are the EP2/EP4 subtypes. Further, we provide evidence of SR-BI and ABCA1 induction by aspirin treatment.
1. Introduction
In atherosclerosis, as in other inflammatory diseases, macrophages are the most predominant leucocytes in the lesion [1] where they contribute to the process by releasing mediators, turning into foam cells and phagocytosing apoptotic cells [2].
The macrophage scavenger receptor CD36, which binds native and modified lipoproteins, has been implicated in the formation of foam cells [3] but also in the phagocytosis of apoptotic cells during atherogenesis [4]. Corticosteroids, TGF-β1 and LPS downregulate CD36, while M-CSF, GM-CSF, native and modified LDL, cellular cholesterol and IL-4 upregulate it, consistent with the hypothesis that CD36 expression correlates with monocyte maturation and is downregulated during inflammation [5]. In this regard, the peroxisome proliferator-activated-receptor (PPAR) γ, which has anti-inflammatory properties, is an activator of CD36 expression [6].
PPAR (α, β/δ and γ) are ligand-activated transcription factors that control lipid and glucose homeostasis. They are members of the nuclear receptor family and form heterodimers with the retinoid X receptor [7]. PPARs are activated by fatty acids and fatty acid-derived compounds (such as some eicosanoids) [8,9]. In addition, PPARα is activated by fibrates [8] and PPARγ binds the antidiabetic glitazones [10]. Both PPAR α and γ are expressed, among other cell types, in differentiated human macrophages [11]. The function of PPARβ/δ is relatively unknown although recent reports have implicated it in the same processes as PPAR α and γ, such as inflammation [12], lipid transport and metabolism, and macrophage differentiation. Several studies have repeatedly suggested the importance of PPARs as modulators of atherosclerosis development [11].
Additionally, atherosclerosis is associated with an increase in prostaglandin biosynthesis [13]. Prostaglandins are synthesized from arachidonic acid by cyclooxygenase (COX), which has two isoforms, COX1 and COX2. Nonsteroidal anti-inflammatory drugs (NSAIDS) inhibit both isoforms and, therefore, block the inflammatory prostaglandin elements of the eicosanoid cascade [14]. Recently, PPARγ ligands have been reported to inhibit COX2 transcription in macrophages [15] in addition to inducing CD36 [6,15,16]. Therefore, CD36 might be regulated by a mechanism depending on prostaglandin synthesis. More specifically, acetylsalicylic acid (aspirin, ASA), an NSAID that is widely used in patients with atherosclerosis, could regulate CD36 by reducing prostaglandin synthesis [14].
In the present study, we show that, at therapeutic plasma concentrations [14,17], aspirin increases CD36 expression in human THP-1 macrophages. We also provide data indicating that this induction is PGE2 dependent and PPARγ independent. We also studied the effect of aspirin on SR-BI and ABCA1, which are important as lipoprotein receptors in atherosclerosis development. SR-BI is a scavenger receptor class B type I that binds native and modified lipoproteins [18] and is a physiologically relevant HDL receptor [19,20] that mediates selective lipid uptake [21]. In addition, it also mediates cholesterol efflux [22] and binds to phospholipids, including those confined to the outer layer of apoptotic cells [23]. SR-BI has a protective effect on atherosclerosis development [24,25]. On the other hand, ABCA1 is an ATP-binding cassette (ABC) transporter that mediates cholesterol and phospholipid transfer to cell surface-bound apolipoproteins [26]. This receptor is defective in Tangier disease patients who show an increase in cardiovascular disease [27]. We also provide evidence that aspirin induces SR-BI and ABCA1.
Modulation of these receptors in macrophages might be an important aspect of the inflammatory response and could have a potential use in the pharmacological treatment of inflammation and atherosclerosis.
2. Methods
2.1. HDL isolation
Lipoproteins were obtained by sequential ultracentrifugation of plasma from human patient donors at the Servicio de Hematología del Hospital Clínico de Barcelona in a Beckman Ty65 rotor at densities between 1.063–1.21g/l (HDL) [28]. Lipoproteins were re-centrifuged, dialyzed, and assayed for protein [29].
2.2. Cell culture
The human monocytic cell line THP-1 (from American Type Culture Collection (ATCC)) was cultured in RPMI 1640 supplemented with l-glutamine (GibcoBRL) penicillin/streptomycin (100 U/100 μg/ml, (GibcoBRL)) and 10% fetal bovine serum (Myoclone super plus FBS USA (GibcoBRL), medium A). Cells were maintained at 37 °C in 5% CO2-95% air in an incubator [30].
To allow monocytes to differentiate to adherent macrophages, THP-1 cells were washed in PBS (calcium- and magnesium-free GibcoBRL, buffer A) and resuspended in fresh medium A containing phorbol 12-myristate–13-acetate (50 ng/ml PMA, Sigma). Cells were plated in 24-well dishes at a density 500,000 cells/well and incubated for 3 days. Macrophages were washed in buffer A and left to stand for 1 day in medium A [31].
2.3. Cell viability analysis (MTT assay)
After treatment, 100 μl of 3-(4,5-dimethyl-thiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT, 5 mg/ml PBS) was added to each well, which contained 1 ml of treatment medium. After incubation for 3 h at 37 °C, the medium was aspirated. The formazan crystals were then dissolved by adding 1 ml of 100g/l SDS and 100 mM acetic acid in DMSO.
Aliquots from each well were read on a microELISA plate reader at 570 nm (Bio-Rad 550). The reading is directly proportional to cell viability and results are expressed as percentage of cell viability with respect to control absorbance (100% cell viability).
2.4. Western blot
Cell monolayers were washed in PBS and lysed with 1% Triton/PBS. Total protein was separated by SDS–polyacrylamide gel electrophoresis on 10% polyacrylamide gels with a mini-PROTEAN II dual slab cell (Bio Rad). Proteins from polyacrylamide gels were blotted onto nitrocellulose membranes at 40 mA for 1 h at 4 °C. The residual binding capacity of the membranes was blocked with 5% nonfat milk in PBS (pH 7.5), 0.1% Tween 20. Blots were incubated with monoclonal antibodies against SR-BI (Novus Biologicals) and ABCA1 (Novus Biologicals) or with a monoclonal anti-β-actin (clone AC-15, Sigma). Bound antibody was detected by using the appropriate horseradish peroxidase conjugated antibody. Signals were detected with the ECL on a standard X-ray system [32].
2.5. Flow cytometric analysis
Macrophages were washed in RPMI 1640 containing penicillin–streptomycin (medium B) and treated with aspirin in the same buffer at the indicated concentrations and times. The pH was not altered by aspirin. The various agonists–antagonists were added 30 min after the aspirin. After incubation, macrophages were washed in buffer A and further incubated in 0.5 ml 0.5% BSA-FAF (Sigma)/2 mM EDTA/buffer A (buffer B) for 30 min. Cells were resuspended by pipetting and then centrifuged at 1000 × g at 4 °C for 10 min. Supernatants were discarded by decantation and pellets were resuspended in 20 μl of PE (phycoerythrin) anti-human CD36 antibody (Pharmingen). After 30 min on ice in the dark, cell pellets were washed once in 1 ml buffer B and then centrifuged at 1000 × g at 4 °C for 10 min, supernatants were discarded and pellets were resuspended in 0.5 ml buffer B. Cell associated fluorescence was determined by using a flow cytometer (Epics XL). Flow cytometry experiments were evaluated on 8000 individual cells gated for macrophages based on their forward and side light-scatter profiles. The mean fluorescence intensity was used to represent the data.
For HDL binding assay, treated macrophages were washed twice in RPMI and 10 μg protein HDL/ml RPMI labeled with Alexa 488 (Molecular Probes Alexa 488 protein labeling kit) was added to each well. After 2-h incubations, cells were detached by trypsin–EDTA treatment and transferred to 0.5 ml of RPMI 0.5% BSA-FAF and held on ice. Immediately prior to flow cytometric analysis, the cells were pelleted at 500 × g for 2 min and resuspended in PBS 0.5% BSA-FAF. Alexa-HDL binding was measured as described elsewhere [33].
2.6. Statistics
Data are presented as mean ± standard deviation of at least three experiments. An analysis of variance (ANOVA), combined with the Student–Newman–Keuls test, was used to evaluate the statistical significance of the differences. The computer programme Statview was used for the calculations.
3. Results
3.1. CD36 is increased by aspirin
Aspirin increased CD36 expression levels in THP-1 macrophages, in a dose- (Fig. 1A,B) and time-dependent manner (Fig. 1C). At 300 μM of aspirin, CD36 increase was significant (p<0.05) at 45 h (Fig. 1C). There was no difference in cell viability between treated and control cells at any time or concentration of aspirin tested (Fig. 2A,B).
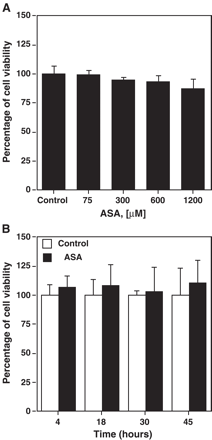
Cell viability in the presence of aspirin. Cell viability at different ASA concentrations (A) or at ASA 300 μM at different time points (B) was determined by the MTT technique and expressed as percentage of absorbance. Data represent mean ± SD of at least three experiments run in triplicates.
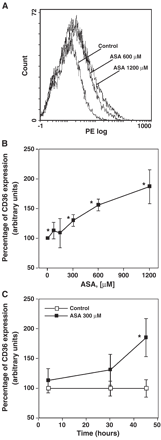
Induction of CD36 expression on macrophages by aspirin (ASA). Macrophages were stimulated with different concentrations of ASA for 45 h (A and B) or with 300 μM of ASA at different time points (C) and CD36 expression was analyzed by incubating PE (phycoerythryn) labeled anti-human CD36 antibody and determining cell fluorescence by flow cytometry. (A) A representative flow cytometry diagram, the mean fluorescence intensity in the control untreated macrophages was 1.1, while ASA 600 μM increased the mean fluorescence to 1.6 and ASA 1200 μM to 2.0. (B) The mean intensities obtained with different ASA concentrations are represented as percentages of CD36 expression. (C) The mean intensities obtained with 300 μM ASA at different time points are represented as percentages of CD36 expression. Data represent mean ± SD of at least three experiments run in triplicates. *p<0.05.
3.2. CD36 induction by aspirin is reversed by PGE2
CD36 induction by aspirin could be indirectly due to a decreased concentration of some prostaglandins but also due to the additional actions that aspirin possess besides its ability to inhibit cyclooxygenase. In fact, it has been shown that some NSAIDs display properties as PPAR agonists [34], which are CD36 modulators [35–37]. Thus, we wanted to study the potential effect of prostacyclin and the mainly released prostaglandins or their metabolites together with the possible PPAR agonism of aspirin on CD36 induction in macrophages. To this purpose, carbaprostacyclin (cPGI2), an analogue of PGI2 which can act as a PPARα/β agonist, 15-deoxy-Δ [12,14]-prostaglandin J2 (15-d-PGJ2), a PGD2 metabolite which can act as a PPARγ agonist, and PGE2, which lacks any activity on PPARs [11], were evaluated to see whether they could revert the CD36 induction by aspirin.
cPGI2 (at both 0.5 and 5 μM) increased expression of CD36 (Fig. 3A). In the presence of 600 μM of aspirin, CD36 induction by cPGI2 was reduced to the CD36 levels produced by aspirin alone. In contrast, addition of 300 μM of aspirin to cells incubated with cPGI2 did not modify the CD36 induction (Fig. 3A).
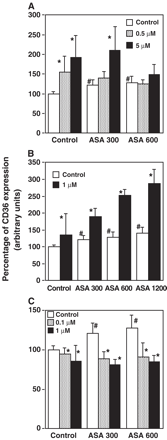
Effect of cPGI2 (5 and 0.5 μM), 15-d-PGJ2 (1 μM) and PGE2 (1 and 0.1 μM) on CD36 expression. cPGI2 (A), 15-d-PGJ2 (B) or PGE2 (C) was added to 30′ ASA treated macrophages. After 45 h, CD36 expression was analyzed by incubating PE labeled antihuman CD36 antibody and determining cell fluorescence by flow cytometry. CD36 expression is represented as the percentage of mean fluorescence intensity. Data represent mean ± SD of at least three experiments run in triplicates. #p<0.05 compared to the corresponding non ASA treated cell control; *p<0.05 compared to the corresponding non prostaglandin treated cell control.
15-d-PGJ2 (1 μM) also induced high levels of CD36 (Fig. 3B). Addition of 15-d-PGJ2 to macrophages treated with different concentrations of aspirin produced a synergic increment of CD36 expression (Fig. 3B).
PGE2 (0.1 and 1 μM) reduced CD36 expression (Fig. 3C). Addition of PGE2 (0.1 and 1 μM) to macrophages treated with 300 or 600 μM of aspirin reduced CD36 to basal levels. These data suggest that the induction of aspirin could be at least partially mediated by inhibiting PGE2 synthesis.
To further confirm the involvement of prostaglandin inhibition on the induction of CD36, we tested the effect of salicylic acid, an analog of aspirin that is a much weaker inhibitor of prostaglandin formation [38]. As it is shown in Fig. 4, salicylic acid (1200 μM) did not affect CD36 expression, while aspirin at the same concentration increased it by 45%.
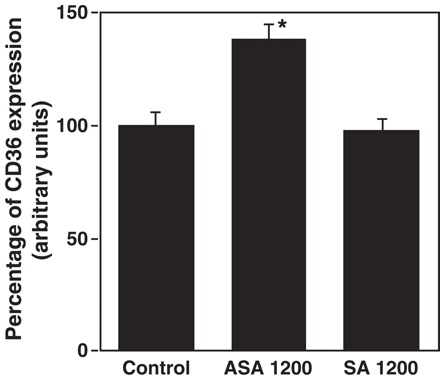
Effect of salicylic acid (SA) on CD36 expression. Macrophages were stimulated with 1200 μM of ASA or SA for 45 h and CD36 expression was analyzed by incubating PE labeled anti-human CD36 antibody and determining cell fluorescence by flow cytometry. CD36 expression is represented as the percentage of mean fluorescence intensity. Data represent mean ± SD of at least three experiments run in triplicates. *p<0.05 compared to the control.
3.3. CD36 induction produced by aspirin is not reversed by PPARγ antagonists
Although previous reports had shown that aspirin acts by a PPARγ independent mechanism [34,39], in our conditions we could not rule out a possible involvement of this nuclear receptor in mediating its effects on CD36. Therefore, we measured the effect of two PPARγ antagonists, diclofenac, which is also a cycloxygenase inhibitor [40], and BADGE [41] on the induction of CD36 expression by aspirin.
BADGE did not reverse CD36 induction in macrophages treated with 1200 μM aspirin (Fig. 5). In contrast, diclofenac increased CD36 expression by itself and further increased it in aspirin-treated macrophages. These results suggest that aspirin induces CD36 by a PPARγ independent mechanism.
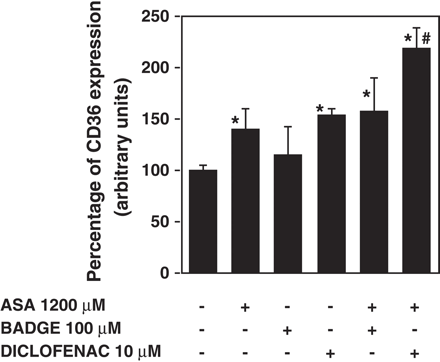
Effect of PPARγ antagonists, BADGE (100 μM) and diclofenac (10 μM) on CD36 induction by aspirin (ASA). Diclofenac or BADGE was added to 30′ ASA treated macrophages. After 45 h, CD36 expression was analyzed by incubating PE labeled antihuman CD36 antibody and determining cell fluorescence by flow cytometry. CD36 expression is represented as the percentage of mean fluorescence intensity. Data represent mean ± SD of at least three experiments run in triplicates. *p<0.05 when compared to the non ASA treated cell control; #p<0.05 when compared to ASA treated cells.
3.4. CD36 induction produced by aspirin is reversed by EP2 and EP2/EP4 subtype EP (E-type prostaglandin) receptor agonists
Since a reduction of the production of PGE2 could be at least in part mediating the effect of aspirin on CD36 expression, we wanted to study the role of the different EP receptor subtypes on CD36 induction by aspirin. Four subtypes of EP receptors (EP1, EP2, EP3, and EP4) from various species were previously defined pharmacologically and have recently been cloned [42]. Activation of EP1 receptors is associated with increases in intracellular Ca2+; EP2 and EP4 lead to increased levels in intracellular cAMP and EP3 reduces intracellular cAMP levels [43]. We tested thapsigargin, an inducer of intracellular Ca2+, which would mimic the effect of EP1 activation, and different EP receptor agonists, which act at different EP receptor subtypes.
Thapsigargin 10 nM did not reverse CD36 aspirin induction (Fig. 6A). In addition, sulprostone (0.2 and 2 μM, Fig. 6B), an agonist of EP1/EP3 receptors [43,44], did not alter the effect of aspirin on CD36, thus suggesting that neither EP1 nor EP3 was involved. On the other hand, butaprost, an EP2 agonist [45,46], and prostaglandin E1 alcohol, an EP2/EP4 agonist, [47] diminished or abolished, respectively, CD36 induction by aspirin (Fig. 6C,D). The inhibition of PGE1 alcohol on CD36 expression induced by aspirin was complete even at 0.01 μM. In contrast, butaprost did not fully inhibit CD36 expression even at 50 μM (Fig. 6D). Taken together these data suggest that aspirin reduces PGE2 synthesis, which inhibits CD36 expression by acting through EP2 and EP4 receptors.
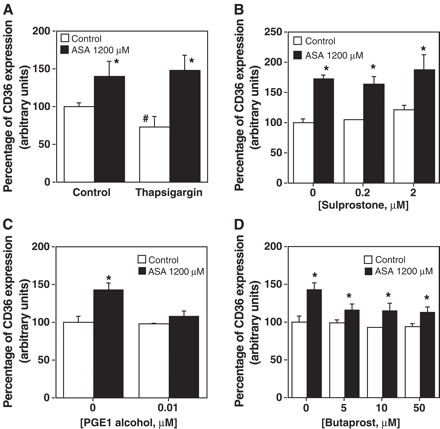
Effect of thapsigargin and different EP receptor subtype agonists on CD36 induction produced by aspirin. Thapsigargin at 10 nM (A), sulprostone (B), PGE1 alcohol (C) or butaprost (D) was added to 30′ ASA treated macrophages. After 45 h, CD36 expression was analyzed by incubating PE labeled antihuman CD36 antibody and determining cell fluorescence by flow cytometry. CD36 expression is represented as the percentage of mean fluorescence intensity. Data represent mean ± SD of at least three experiments run in triplicates. The statistical significance is indicated with * and # and p is at least <0.05 when compared to the non ASA treated cell control.
3.5. Aspirin treatment increases HDL binding to macrophages
CD36 can function as an HDL receptor [5]. To examine the effect of aspirin on the interaction of HDL with its receptors, we analyzed HDL binding by incubation of macrophages with Alexa-488-labeled HDL. Fluorescence incorporated to macrophages was analyzed by flow cytometry. Alexa-HDL (10 μg/mL) binding increased with aspirin treatment of macrophages at 45 h (Fig. 7).
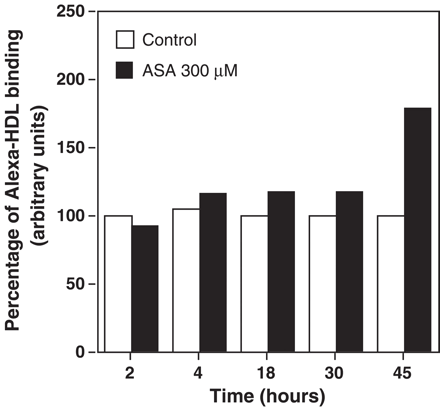
Effect of aspirin (ASA) on Alexa-HDL percent binding to macrophages. Macrophages were stimulated with ASA (300 μM) at different time points after which macrophages were incubated with Alexa-HDL (10 μg protein HDL/mL) for 2 h. Cells were then treated as described in the Methods section. HDL binding is represented as the percentage of mean fluorescence intensity. Data represent mean of two experiments run in triplicates.
3.6. Aspirin increases SR-BI and ABCA-1 expression
As the increase in HDL binding could also be due to the induction of SR-BI, its expression was analyzed. ABCA1 expression in response to aspirin was also studied to see the potential effect of aspirin on other HDL related receptors.
A significant induction of SR-BI was observed when macrophages were incubated with 600 and 1200 μM of ASA (p<0.05, Fig. 8). ABCA1 expression was maximally induced with 300 μM and was not further increased with higher concentrations of ASA (300,600 and 1200 μM, p<0.05 vs control, Fig. 8).
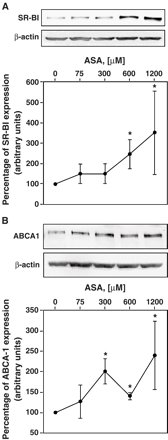
Effect of ASA on SR-BI and ABCA1 expression levels. Macrophages were stimulated with different concentrations of ASA. After 45 h, cells were lysed and expression of SR-BI (A) and ABCA1 (B), respectively, was determined by Western blot as described in Methods. Three different experiments were performed in duplicates. Data are presented as mean ± SD. Representative blots and quantitative analysis are shown. *p<0.05.
4. Discussion
During the process of monocyte differentiation to macrophages in the early stages of atherosclerotic plaque formation, CD36 expression increases [6], which seems to be a key factor in the subsequent formation of foam cells [3]. Moreover, as prostaglandins, products of cyclooxygenase activity, are involved in several stages of atherosclerotic plaque formation [48], they might participate in the regulation of CD36 expression. If so, a cyclooxygenase inhibitor such as aspirin could also modulate CD36 expression. Indeed, all three prostaglandins tested here modify CD36 expression in macrophages and aspirin increases it.
In particular, cPGI2 induced CD36 expression. This induction could be caused by cPGI2 acting as a PPARα/β agonist [11] since these agonists also induce CD36 [35,36]. At 600 μM, aspirin reduced the CD36 induction produced by cPGI2. In addition to its capacity to induce CD36, aspirin, at the highest concentration assayed, may act as an antagonist of PPARα/β, PGI2 receptor, or other cPGI2properties. In support of this hypothesis, it has been shown that some NSAIDs disrupt the DNA binding capacity of PPARβ–RXRα heterodimers [49] and that aspirin reduces PPARα protein [50] in rat liver cells.
15-d-PGJ2, which is also a PPARγ activator, increased CD36 expression (Fig. 3B), as previously reported [51]. The addition of 15-d−PGJ2 to aspirin treated cells further increased CD36 expression. This increase was greater than the sum of the increases produced by these two agonists separately. This potentiation effect indirectly suggests that each agonist uses a different signaling pathway and that aspirin acts on CD36 expression independently of PPARγ. Indeed, neither aspirin nor sodium salicylate activates either PPARα or PPARγ in vitro [34,39]. To confirm the PPARγ independent effect of aspirin, macrophages were incubated with two PPARγ antagonists, BADGE (100 μM) [41] and diclofenac (10 μM) [40], which also inhibits cyclooxygenase. BADGE did not modify CD36 expression induced by aspirin, while diclofenac further increased the induction of CD36 expression produced by aspirin. Most likely this is due to an accumulative effect of both drugs inhibiting cyclooxygenase activity.
The other prostaglandin tested, PGE2, abolished the induction produced by aspirin (Fig. 2C). Our results indicate that the effect of aspirin on CD36 could be in part mediated by a decrease in PGE2 production. This could be effected by at least two complementary mechanisms: (i) a direct inhibitory effect on the COX enzyme activity and (ii) a reduction of cox-2 gene transcription, a COX isoform expressed in basal condition in macrophages [52,53]. The lack of effect of salicylic acid, that is able to reduce COX2 protein levels [54], on CD36 expression, argues against the latter possibility.
PGE2 is considered as one of the most atherogenic eicosanoids [55] predominantly secreted by macrophages [56]. Thus, among the aspirin effects related to the inhibition of prostaglandin production, the inhibition of PGE2 may be the most relevant to explain the induction of CD36 in macrophages. It is tempting to speculate that the beneficial effect of aspirin on atherosclerosis may also be due to inhibition of PGE2 production [57]. One of the positive effects of this inhibition could be to reduce the expression of metaloproteinases 2 and 9, which destabilize the atherosclerotic plaque [58]. However, our results indicate that the inhibition of PGE2 production by aspirin would lead to an increase of CD36 expression, which has been linked to a pro-atherogenic effect. Increased expression of CD36 stimulates oxLDL uptake [3] and, consequently, foam cell formation. In addition, knocking out CD36 in apoE-deficient conditions reduces atherosclerosis formation [59]. Interestingly, increased CD36 expression has also been found in antiatherogenic situations [60]. As other authors have already suggested for the action of some glitazones on macrophages, the antiatherogenic effects of aspirin, such as the induction of SR-BI or ABCA1 found here, may be more relevant than the proatherogenic effect of the increased expression of CD36 [60,61].
It could be argued that the reduction of the symptomatic plaque observed in rheumatoid arthritis patients treated with high doses of aspirin [62], or the beneficial effects of low doses of aspirin in primary and secondary prevention of cardiovascular disease [63] therapy, could be mediated by the mechanisms described here. The relevance and the exact mechanisms by which aspirin affects SR-BI, CD36 and ABCA1 in vitro remain to be determined, as does the role of CD36 expression in antiatherogenic conditions.
Acknowledgements
This work was supported by grants from FPCNL, CICYT (SAF03/01232 and SAF2004-03045), European Union FEDER founds, FIS Red Temática G03/181, “Fundación Ramón Areces” and Generalitat de Catalunya (2001SGR00141). MV was a recipient of Reincorporació de doctors de la Generalitat de Catalunya (RED). GL was supported by a grant of the University of Barcelona. We thank Mr. R. Rycroft (Language Advice Service of the University of Barcelona) for helpful assistance.
References
Author notes
Time for primary review 26 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}