





Time for primary review: 15 days
Chaperone-mediated autophagy (CMA) is a selective mechanism for the degradation of soluble cytosolic proteins bearing the sequence KFERQ. These proteins are targeted by chaperones and delivered to lysosomes where they are translocated into the lysosomal lumen and degraded via the lysosome-associated membrane protein type 2A (LAMP-2A). Mutations in LAMP2 that inhibit autophagy result in Danon disease characterized by hypertrophic cardiomyopathy. The ryanodine receptor type 2 (RyR2) plays a key role in cardiomyocyte excitation–contraction and its dysfunction can lead to cardiac failure. Whether RyR2 is degraded by CMA is unknown.
To induce CMA, cultured neonatal rat cardiomyocytes were treated with geldanamycin (GA) to promote protein degradation through this pathway. GA increased LAMP-2A levels together with its redistribution and colocalization with Hsc70 in the perinuclear region, changes indicative of CMA activation. The inhibition of lysosomes but not proteasomes prevented the loss of RyR2. The recovery of RyR2 content after incubation with GA by siRNA targeting LAMP-2A suggests that RyR2 is degraded via CMA. In silico analysis also revealed that the RyR2 sequence harbours six KFERQ motifs which are required for the recognition Hsc70 and its degradation via CMA. Our data suggest that presenilins are involved in RyR2 degradation by CMA.
These findings are consistent with a model in which oxidative damage of the RyR2 targets it for turnover by presenilins and CMA, which could lead to removal of damaged or leaky RyR2 channels.
1. Introduction
Ryanodine receptor type 2 (RyR2) is the major cardiac sarcoplasmic reticulum (SR) calcium-release channel. RyR2 opens in response to calcium influx via the L-type calcium channel, leading to release of large quantities of calcium required for contraction of the heart. For relaxation, RyR2 must close to allow the re-uptake of calcium into the SR by the Ca2+-ATPase.1 Impairments in this cycle of release and re–uptake of SR calcium cause cardiac contractile dysfunction. The SR is severely compromised in myocardial ischaemia.2 Proteolysis of SR calcium handling proteins occurs during ischaemia–reperfusion (I/R).3–6 RyR2 levels are especially vulnerable to ischaemic injury and decline early in ischaemia.7 Cardiomyocytes that survive an episode of I/R show a 50–70% decrease in RyR2 levels. This decrease in RyR2 may be cardioprotective as siRNA depletion of RyR2 promotes survival of cardiomyocytes from simulated I/R injury.8 In fact, turnover of damaged RyR2 may play a protective role in vivo, as leaky RyR2 channels have been suggested to contribute to myocardial remodelling after I/R.9
We recently demonstrated that RyR2 can be degraded by the concerted action of calpains and proteasomes during simulated I/R (sI/R) in cultured cardiomyocytes.10 Surprisingly, inhibition of macroautophagy did not prevent RyR2 degradation during sI/R and but instead stimulated an even greater degradation of this protein. It has been shown that inhibition of macroautophagy can stimulate a compensatory activation of chaperone-mediated autophagy (CMA).11 CMA is a selective autophagic mechanism for the degradation of soluble proteins.12 For degradation via CMA, cytoplasmic proteins bearing the sequence KFERQ, or a closely related sequence, are targeted by chaperones and delivered to the lysosomes where, after interaction with the lysosome- associated membrane protein type 2A (LAMP-2A), they are translocated into the lysosomal lumen and degraded.12 Reactive oxygen species (ROS) may generate oxidized protein substrates exposing the KFERQ sequence recognized by CMA-specific chaperones.13
We demonstrate here that exposure of cultured cardiomyocytes to oxidative stress triggers the degradation of RyR2 by CMA. Further, in silico analysis of the rat RyR2 amino acid sequence revealed six motifs related to KFERQ in the cytoplasmic domain of the protein, supporting RyR2 as a potential target for CMA-dependent proteolysis. Additionally, induction of CMA by geldanamycin (GA) decreased RyR2 levels in a manner dependent on LAMP2, presenilins, and ROS.14 Together, these data suggest a model in which CMA targets damaged RyR2 channels for degradation.
2. Methods
2.1 Primary cultures
All animal experiments were approved by the Animal Care and Use Committee of the University of Chile and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication, 8th Edition, 2011). Neonatal Sprague Dawley rats (1–3 day old) were euthanized by decapitation. Cardiomyocytes were isolated from rat hearts by enzymatic digestion using pancreatin (1.2 mg/mL) and collagenase (0.2 mg/mL) as described previously.15 Cells were pre-plated to discard non-myocyte cells and the myocyte-enriched fraction was plated at 1.0 × 106 cells/mm2 on gelatin-precoated 35 mm dishes (Falcon, BD Biosciences, Oxford, UK) and grown in Dulbecco's modified Eagle medium and M199 medium (DMEM/M199; ratio 4/1), with 10% (w/v) fetal bovine serum (FBS) for 24 h before the experiments. Cardiomyocyte cultures were at least 95% pure as evaluated by immunofluorescence using an anti-β-myosin heavy chain antibody (Vector Laboratories, Burlingame, CA, USA). Cells were grown and maintained at 37°C in an incubator containing 95% O2 and 5% CO2.
Pancreatin, DMEM, M199, clasto-lactacystin-β-lactone (Lac), MG132, 3-methyladenine (3-MA), GA, ammonium chloride (NH4Cl), chloroquine (Clo), N-acetylcysteine (NAC), hydrogen peroxide (H2O2), and ionomycin were purchased from Sigma-Aldrich Corp (St Louis, MO, USA). Collagenase was from Invitrogen (Paisley, Scotland, UK). Fetal bovine serum was from Biological Industries, Kibbutz Beit Haemek, Israel. The γ-secretase inhibitor XXI (Compound E, CE) was from Merck (Whitehouse Station, NJ, USA).
2.2 Experimental protocols
Cultured cardiomyocytes were incubated with 2 µM GA for 6 h. When used Lac (2.5–5 μM), MG132 (10 µM), 3-MA (10 mM), NH4Cl (10 mM), Clo (100 μM), NAC (3 mM), or CE (10 nM) were added 15 min before GA. Small interfering RNA (siRNA) was constructed essentially as described before,16 a 25-nucleotide siRNA 5′-AAGCGCCAUCAUACUGGAUAUGAGC-3′ against rat LAMP-2A mRNA, and a scrambled (Scr) siRNA containing the 25-nucleotide sequence 5′-AAUUUAGCCGAUACUGCCUAGACGA-3′. Cells were grown in 35 mm dishes and, after 24 h, transfected with siRNAs at a concentration of 25 nM with oligofectamine (Invitrogen, Carlsbad, CA, USA). After overnight incubation, the medium was removed and replaced with 10% FBS in DMEM/M199. LAMP-2A down-regulation was assessed 24 and 48 h post-transfection.
2.3 Protein extraction and western blot analysis
Cardiomyocytes were harvested, washed with ice-cold PBS to eliminate dead cells and homogenized in cold lysis buffer (CytoBuster™ Protein Extraction Reagent, Novagen Inc., Madison, WI, USA) in the presence of a protease inhibitor cocktail (Complete, Roche Diagnostics, Mannheim, Germany). The lysate was centrifuged at 10 000g for 10 min at 4°C and the supernatant was collected. Proteins were separated by SDS–PAGE (3.5–8% gradient gels for RyR2, 15% gels for LC3-II, or 8% gels for LAMP-2A and α-spectrin). After electrophoresis, proteins were transferred onto PVDF membranes (Millipore Corp., Bedford, MA, USA). The primary antibodies used were: anti-RyR2 (Affinity BioReagents Inc., Golden, CO), anti-LAMP-2A (Zymed laboratories, South San Francisco, CA, USA), and anti-LC3B (Cell Signaling Technology, Danvers, MA, USA), anti-α-spectrin (Millipore Corp., Bedford, MA, USA), and anti-β-actin (Sigma-Aldrich Corp., St Louis, MO). After incubation with the appropriate secondary antibody, antigen–antibody reaction was detected by ECL (Amersham Biosciences, Pittsburgh, PA, USA) or Odyssey Licor and quantified by densitometric analysis with Quantity One (BioRad, Hercules, CA, USA) or Odyssey Licor (version 3.0) imaging system. Results were normalized with respect to β-actin.
2.4 Immunocytochemistry
Cells were plated on coverslips in a 35 mm plate to 50% confluence and 48 h later they were stimulated with GA (2 µM) for 6 h. Cells were carefully washed with PBS and fixed in a 4% paraformaldehyde for 20 min. Fixed cells were washed and permeabilized with PBS-Triton (0.1% Triton X-100 in PBS) for 10 min at room temperature, blocked with 3% BSA for 1 h, and incubated overnight at 4°C with anti-LAMP-2A (1:500) and anti-Hsc-70 (1:1000, Abcam Inc., Cambridge, MA, USA) in 3% BSA. Cells were then washed with PBS and incubated at room temperature for 1 h with secondary antibodies. Alexa Fluor 488 anti-rabbit IgG (1:300, Invitrogen, Carlsbad, CA, USA) was used for LAMP-2A and Alexa Fluor 633 anti-mouse IgM was used for Hsc-70 (1:300, Invitrogen, Carlsbad, CA, USA). Stained cells were mounted with DAKO fluorescent mounting media and the images were captured with a LeicaTCSSP1 confocal microscope workstation (Leica Microsystems, Wetzlar, Germany). Images were analysed with the software ImageJ (NIH, Bethesda, MD, USA).
2.5 Cell viability
The effect of GA on cell viability was determined by the ability of the cell membrane to exclude propidium iodide (PI). After incubation with GA, cells were trypsinized, collected along with the culture medium, and the incorporation of PI was quantified in an FACScan flow cytometer.10 Additionally, viability was assessed using a CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI, USA) to measure lactate dehydrogenase in the cultured medium after 6 h treatment with GA.
2.6 Other procedures
Oxidative stress induced by GA was determined by measuring ROS generation by flow cytometry using dihydrorhodamine 123 (Sigma-Aldrich Corp, St Louis, MO). Proteins were quantified according to Hartree.17 Cultured cardiomyocytes were incubated with H2O2 (100 µM) for 2 or 6 h. Simulated ischaemia/reperfusion (sI/R) was performed essentially as described before,10 with the timing of 6 h ischaemia and 12 h reperfusion.
2.7 Statistical analysis
Data are shown as mean ± SEM of the indicated number (n) of independent experiments. Data were analysed by Student's t-test or one-way ANOVA followed by Tukey's test. Differences were considered significant at P < 0.05.
3. Results
3.1 Oxidative stress-induced declines in steady-state RyR2 levels are inhibited by blocking lysosomal processing
I/R injury results in a decrease in RyR2 receptor levels. We recently demonstrated that in cultured cardiomyocytes, the decrease in RyR2 after I/R could be inhibited by blocking the proteolytic processing of either calpains or the proteasome.10 Macroautophagy was not implicated, as treatment with 3-MA resulted in an even greater reduction of RyR2 levels. As inhibition of macroautophagy has been shown to result in the stimulation of CMA,11 we set out here to examine the effect of lysosomal inhibition on RyR2 degradation.
Cultured cardiomyocytes were treated with 100 µM H2O2 for 6 h in the presence or absence of NH4Cl (to block lysosomal proteolysis) or MG132 (to block proteasomal proteolysis). Treatment of cardiomyocytes with H2O2 resulted in a >50% reduction in the steady-state levels of RyR2. This reduction was blocked by the inhibition of both lysosomal and proteasomal proteolysis. The inhibition of both degradation pathways together further increased the steady-state levels of RyR2 (Figure 1A). These data suggest a role for lysosomal proteolysis in RyR2 degradation and are consistent with CMA-dependent processing of the RyR2.
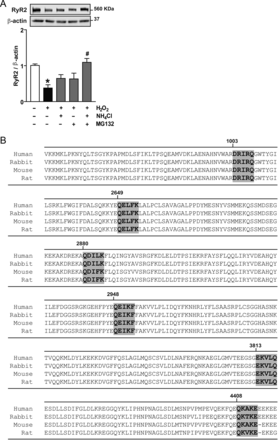
(A) The degradation of RyR2 is stimulated by hydrogen peroxide (H2O2). Representative western blot against RyR2 or β-actin in cultured cardiomyocytes treated with H2O2 in the presence or absence of NH4Cl, MG132, or NH4Cl and MG132. The bar graph shows the ratio of RyR2/β-actin. (B) Motifs biochemically related to the pentapeptide KFERQ (box) in the RyR2 aminoacidic sequence from Rattus novergicus (GenBank ID: NP_114467.1), human (GenBank ID: NP_001026.2), rabbit (GenBank ID: NP_001076226.1), and mouse (GenBank ID: NP_076357.2). Values are mean ± SEM (n = 5–6). *P < 0.05 H2O2 vs. control and #P < 0.05 H2O2 vs. H2O2 plus NH4Cl and MG132.
3.2 Rat RyR2 sequence harbours six sequences related to KFERQ
The specificity of degradation by CMA appears to require a KFERQ-like motif within the target protein.18In silico analysis of the rat RyR2 amino acid sequence (Figure 1B) revealed six sequence motifs related to KFERQ (GenBank ID: NP_114467.1). These sequences are conserved in human (NP_001026.2), rabbit (NP_001076226.1), and mouse (NP_076357.2) RyR2 amino acid sequence. All these sequences are localized to the cytoplasmic domain of the protein. Thus, RyR2 is a potential target for CMA-dependent proteolysis.
3.3 Geldanamycin activates chaperone-mediated autophagy and stimulates RyR2 degradation
We have shown previously that inhibiting lysosomal processing can block turnover of RyR2. Given this, we reasoned that inducing CMA would have the opposite effect and decrease the steady-state levels of RyR2. Nutrient deprivation and stimulation with GA have been shown to induce CMA in cultured fibroblasts.14 Increases in the cellular content of LAMP-2A and the perinuclear redistribution of lysosomes harbouring membrane-associated LAMP-2A and lumenal Hsc-70 are hallmarks of CMA activation.19,20 To determine whether GA also induces CMA in cultured cardiomyocytes, we evaluated the content and localization of LAMP-2A in cells exposed to 2 µM GA for 6 h. As shown in Figure 2, GA produced a significant increase in both LAMP-2A protein content (Figure 2A) and in its colocalization with Hsc70 around the cell nucleus (Figure 2B). All this data together indicate that, as in fibroblasts, GA activates CMA in cultured cardiomyocytes.
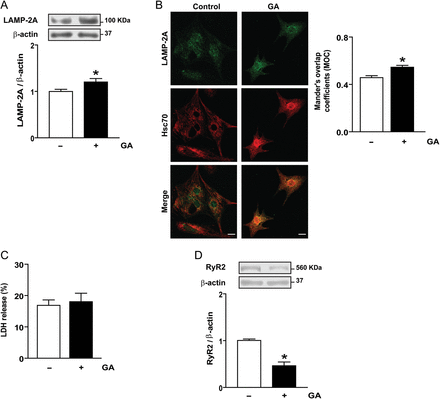
Geldanamycin (GA) stimulates chaperone-mediated autophagy and RyR2 degradation in cultured cardiomyocytes. (A) Representative western blot obtained with anti-LAMP-2A or anti-β-actin in total cell lysates. The bar graph shows the ratio LAMP–2A/β-actin calculated from densitometric analysis of western blot. (B) Colocalization of LAMP-2A and Hsc70 in cells stimulated with GA. Bar graph indicated Mander's coefficients of the overlap. The scale bar is 10 µm. (C) Percent of cell death measured by LDH release is indicated in the bar graph. (D) Representative western blot obtained with anti-RyR2 or anti-β-actin in total cell lysates. The bar graph shows the ratio RyR2/β-actin calculated from densitometric analysis of western blot. Values are mean ± SEM (n = 9 independent experiments). *P < 0.05 GA vs. control.
GA did not alter the morphology or viability of cardiomyocytes measured by LDH (Figure 2C) or propidium iodide (data not shown), but triggered a significant (50% ± 0.073, P< 0.0001) decrease in RyR2 content (Figure 2D) suggesting that GA induced RyR2 degradation. As CMA degrades soluble cytoplasmic proteins, we investigated first whether another proteolytic pathway, activated in addition to CMA during incubation with GA, was responsible for RyR2 degradation.
3.4 Proteasome or macroautophagy are not responsible for geldanamycin-induced RyR2 degradation
We have previously shown that RyR2 is degraded by the proteasome during sI/R in cultured cardiac myocytes.10 To investigate whether the ubiquitin–proteasome system is responsible for RyR2 degradation after GA exposure, cardiomyocytes were incubated with GA in the presence of the proteasome inhibitor Lac (2.5 μM). We found that inhibition of the proteasome during incubation with GA did not prevent the decrease in RyR2 content (Figure 3A), suggesting that RyR2 was not degraded by the proteasome upon incubation with GA. Increasing Lac concentration to 5 µM did not increase RyR2 levels (data not shown).
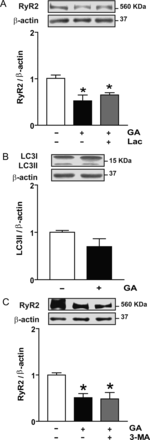
Effect of proteasome or macroautophagy inhibitors on RyR2 content in cardiomyocytes treated with geldanamycin (GA). (A) Representative western blot obtained with anti-RyR2 or anti-β-actin in total cell lysates obtained from controls or after incubation with GA or GA plus the proteasome inhibitor clasto-lactacystin-β-lactone (Lac). The bar graph shows the ratio RyR2/β-actin calculated from densitometric analysis of western blot. (B) Effect of GA on LC3-II content after incubation with GA. Representative western blot obtained with anti-LC3B or anti-β-actin. The bar graph shows the ratio LC3–II/β-actin calculated from densitometric analysis. (C) Effect of 3-methyladenine (3-MA) on RyR2 content after incubation with GA. Representative western blot obtained with anti-RyR2 or anti-β-actin in total cell lysates obtained from controls or after incubation with GA or GA and 3-MA. Bars show the ratio RyR2/β-actin calculated from densitometric analysis of western blot. Values are mean ± SEM (n = 3–9). *P < 0.05 GA, GA plus Lac, or GA plus 3-MA vs. control.
Macroautophagy, typically referred to as autophagy, is a major lysosomal pathway for the degradation of proteins and damaged organelles. Activation of autophagy is characterized by an increase in LC3II, a protein involved in the formation of autophagosomes.21 As shown in Figure 3B, no differences in LC3II content were observed in cardiac myocytes treated with GA compared with controls, suggesting that GA did not trigger autophagy. It is unlikely, therefore, that RyR2 is degraded through this pathway. Further, exposure of cardiomyocytes to GA in the presence of the autophagy inhibitor 3-MA did not prevent the decrease in RyR2, confirming that autophagy was not involved in GA-induced RyR2 degradation (Figure 3C).
3.5 Chaperone-mediated autophagy degrades RyR2 in cardiomyocytes treated with GA
Next, we investigated whether lysosomes are involved in RyR2 degradation by exposing cardiomyocytes to GA in the presence of the lysosome inhibitors ammonium chloride (NH4Cl) or chloroquine (Clo).22 As shown in Figure 4A, incubation with NH4Cl or Clo completely prevented the RyR2 degradation induced by GA, suggesting significant involvement of lysosomal pathways in RyR2 degradation. Incubation with NH4Cl or Clo alone provoked no significant change in RyR2 levels with respect to control.
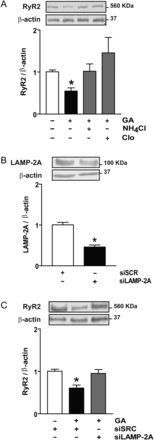
Chaperone-mediated autophagy degrades RyR2 in cardiomyocytes stimulated with geldanamycin (GA). (A) Representative western blot obtained with anti-RyR2 or anti-β-actin from controls, GA, GA plus ammonium chloride (NH4Cl), or chloroquine (Clo) to inhibit lysosomes. Bar graph shows the ratio RyR2/β-actin. (B) Representative western blot against LAMP-2A or β-actin in cardiomyocytes transfected with siRNA for LAMP-2A (siLAMP-2A) or scrambled siRNA (siSCR). Bar graph shows the ratio LAMP-2A/β-actin. (C) Western blot against RyR2 or β-actin obtained in the same cells. Bars show the ratio RyR2/β-actin. Values are mean ± SEM (n = 3–5). *P < 0.05 GA vs. control; siLAMP-2A vs. siSCR; or GA plus siSCR vs. siSCR.
The limiting step in the degradation of a protein through CMA is its interaction with LAMP-2A in the lysosomal membrane.12 In light of this, we transfected cardiomyocytes with siRNA targeting LAMP-2A. After 48 h, LAMP-2A was reduced to 40% of the level observed in cells transfected with a scrambled siRNA (Scr) (Figure 4B), without changes in the content of LAMP-2C, another lysosome-associated membrane protein not involved in CMA (data not shown). The decrease in LAMP-2A expression prevented the decrease in RyR2 induced by GA (Figure 4C), suggesting that CMA is the degradation pathway for RyR2 under this condition.
3.6 The oxidative stress produced by GA triggers the degradation of RyR2 via chaperone-mediated autophagy
GA harbours a benzoquinone moiety that generates ROS.23 GA also reacts non-enzymatically with glutathione, depleting the cell of one of its main redox buffer components,24 and further contributing to the oxidative stress generated by GA. Given this, we set out to test the possible role of ROS accumulation of GA-induced RyR2 degradation.
First, we found that the exposure of cardiomyocytes to GA (2 h) produced a two-fold increase in intracellular ROS, as quantified by flow cytometry with dihydrorhodamine 123 (see Supplementary material online, Figure S1). This increase in ROS levels was maintained after 6 h of exposure (Figure 5A), indicating that GA produced oxidative stress. Given that we had already linked oxidative stress with increased RyR2 turnover (Figure 1), we examined whether in this context, ROS was required to trigger RyR2 degradation. Cells were treated with GA in the presence or absence of the free-radical scavenger N-acetylcysteine (NAC). Increases in intracellular ROS levels were prevented by NAC (Figure 5A), which also prevented the decrease in RyR2 content induced by GA (Figure 5B). Oxidation of RyR2, therefore, seems to be determinant for its degradation via CMA. On the other hand, NAC did not prevent the increase in LAMP-2A induced by GA (Figure 5C), suggesting that oxidative stress and the activation of CMA are two independent effects of GA and that both are required for the targeted turnover of RyR2 by CMA.
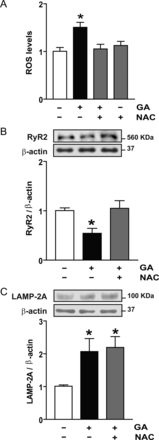
ROS generation and CMA stimulation are two independent effects of geldanamycin (GA). (A) ROS were measured by flow cytometry after 6 h with GA in the presence or absence of N-acetylcysteine (NAC). (B) Representative western blot obtained with anti-RyR2 or anti-β-actin in total cell lysates obtained from controls or after incubation with GA or GA plus NAC. Bar graph shows the ratio RyR2/β-actin. (C) Representative western blot obtained with anti-LAMP-2A or anti-β-actin in total cell lysates. The bar graph shows the ratio LAMP-2A/β-actin calculated from densitometric analysis of western blot. Values are mean ± SEM (n = 5–7). *P < 0.05 GA vs. control or GA plus NAC vs. control.
3.7 Presenilins are involved in RyR2 degradation
We have shown previously that calpains are involved in RyR2 degradation during sI/R in cultured cardiomyocytes.10 To investigate whether these proteases are activated in cardiomyocytes exposed to GA (6 h), we tested for the characteristic appearance of two proteolytic fragments of α-spectrin generated by calpains.25 The results suggested that GA does not activate calpains (Figure 6A).
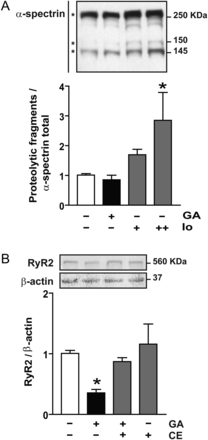
Presenilins are implicated in RyR2 degradation in cardiomyocytes treated with geldanamycin (GA). (A) Western blot representative of α-spectrin proteolysis. Asterisk indicates α-spectrin total (top) or calpain-induced proteolytic fragments (150 and 145 KDa) of α-spectrin. Ionomycin was used as positive control (Io, 0.5+ and 1++ µM). Bar graph shows the ratio within proteolytic fragments 145 KDa and total α-spectrin. (B) Representative western blot obtained with anti-RyR2 or anti-β-actin in total cell lysates from controls or after incubation with GA in the presence or absence of compound E (CE). Bar graph shows the ratio within RyR2/β-actin. Values are mean ± SEM (n = 4). *P < 0.05 GA or Io vs. control.
We next investigated whether presenilins, intramembrane proteases, might be involved in the cleavage of RyR2 from the SR membrane prior to degradation by CMA. Figure 6B shows that compound E, a specific inhibitor of presenilins, completely prevented RyR2 degradation induced by GA treatment. These data, then, suggest that CMA-dependent degradation of RyR2 requires cleavage by presenilins, which we speculate may allow for release of RyR2 from the SR membrane.
3.8 Chaperone-mediated autophagy is not involved in RyR2 degradation during simulated ischaemia/reperfusion
We previously have shown that RyR2 degradation during sI/R requires proteasomal processing.10 To determine if CMA is also involved under these conditions, we repeated these studies by transfecting cardiomyocytes with control or LAMP-2A siRNA and exposing them to sI/R. LAMP-2A knockdown appeared to have no effect on the decreased levels of RyR2 under these conditions (see Supplementary material online, Figure S3). These results suggest CMA is not involved in RyR2 degradation during sI/R.
4. Discussion
CMA is a selective lysosomal pathway best characterized in degradation of specific cytoplasmic proteins.18,26 One exception to this rule is the recent demonstration of CMA-dependent degradation of a membrane protein, the epidermal growth factor receptor (EGFR).27 Here, we show that the RyR2, another integral membrane protein, is also degraded via CMA specifically after oxidative damage.
To induce CMA, cultured cardiomyocytes were treated with GA. This natural-occurring antibiotic inhibits the ATPase activity of the heat shock protein 90 (Hsp90), a chaperone that maintains the correct folding and stability of several proteins. Hsp90 inhibition triggers dissociation from its so-called client proteins. GA also generates superoxide anion23,28 and provokes GSH depletion.24 Dissociated Hsp90 client proteins are oxidized and degraded via the proteasome.29 In cultured fibroblasts, GA has been shown to induce CMA and promote protein degradation through this pathway.14
Incubation of cardiomyocytes with GA produced the characteristic increase in LAMP-2A together with its redistribution and colocalization with Hsc70 in the perinuclear region, changes indicative of CMA activation.19 Collectively, these data show that GA also triggers CMA in cultured cardiomyocytes.
Our previous work showed that RyR2 is degraded via the calpain and ubiquitin–proteasome pathway during I/R.10 Here, we show that both lysosomal and proteasome pathways target RyR2 for degradation after oxidative damage, when oxidation was induced with H2O2 (Figure 1). However, inhibition of lysosomes, but not proteasome, prevented the loss of RyR2 protein when cells were treated with GA. Thus, we were able to isolate the lysosomal degradation of RyR2 from the proteolytic degradation under these conditions. RyR2 protein is very susceptible to oxidation,30 indeed we observe that the ROS triggered by GA is necessary for the CMA targeting of the RyR2 protein. Our initial thought was that the oxidative environment during reperfusion injury might also trigger RyR2 oxidation leading to its degradation by CMA during I/R. In fact, inhibition of lysosomal processing had no impact of RyR2 degradation during I/R, suggesting that the calpain/proteasomal pathway is predominant under these conditions (see Supplementary material online, Figure S2). One possible difference, resulting in the discrepancy between RyR2 degradation during I/R or with GA, may lay with the proteases activated under these conditions. While we have previously demonstrated that calpain cleavage of RyR2 is required for degradation during I/R damage, we observe no activation of calpains by GA treatment (Figure 6A). In contrast, presenilin activity is required for degradation of RyR2 through the CMA pathway (Figure 6B). The discrepancy may be due to the type or degree of oxidation difference between these stimuli or to specific activation of the proteases involved.
Recovery of RyR2 content after incubation with GA by siRNA-dependent targeting of LAMP-2A strongly suggests that RyR2 is degraded via CMA. This pathway requires the presence of a cis-acting pentapeptide KFERQ-motif to be recognized by the chaperone Hsc70 that targets the protein to the lysosome. RyR2 has a large extramembrane domain that contains six of these motifs providing the molecular basis for recognition by Hsc70 and degradation via CMA. Oxidative stress is a robust stimulus for the induction of CMA,12 and the oxidation of proteins facilitates their degradation via CMA. Our results indicate that the oxidative stress elicited by GA and the activation of CMA are two independent events, since NAC prevented the increase in ROS generation without preventing the increase in LAMP-2A.
RyR2 contains a number of highly reactive cysteine residues that make it an easily oxidizable protein.30 Irreversible oxidation interferes with the proper gating of the channel,31 and irreversibly oxidized RyR2 must be degraded to avoid uncontrolled calcium release and cellular damage. GA-induced RyR2 degradation is likely to be triggered by oxidation, as it was prevented when ROS were scavenged by NAC. Moreover, treatment of cardiomyocytes with H2O2 also induced RyR2 degradation. We posit that irreversible RyR2 oxidation changes the conformation of the protein exposing otherwise sterically inaccessible KFERQ-like motifs, allow for their recognition by Hsc70. Alternatively, oxidative damage of RyR2 may lead to the generation of new motifs recognized by the Hsc70.13
Our results also suggest that presenilins could be involved in RyR2 degradation by CMA. Presenilins are intramembrane proteases established to cleave an array of substrates, including multipass transmembrane proteins.32 Presenilin-2 and RyR2 co-localize in the heart, and presenilin-2 can regulate RyR2 activity.33 Given this, we hypothesize that under certain conditions, RyR2 could be a substrate for presenilin-induced proteolysis. The cleavage of this site could pull the protein away from the membrane. Similarly, unfolding may expose KFERQ-like motifs that would be recognized by Hsc70, and the excised peptide could be delivered to lysosomes for degradation.
I/R damage results in a significant decrease of RyR2 levels in cardiac cells.7 siRNA knockdown of RyR2 in cardiomyocytes prior to I/R has been shown to increase survival of these cells.8 Data reported here suggest a model in which ROS damage of RyR2 targets it for turnover by presenilins and CMA. This may be a pathway to remove damaged or leaky channels. Given the role of LAMP-2A in CMA, these results also open the possibility of a causal role for RyR2 receptor dysregulation in the pathophysiology of Danon disease. Additionally, inhibition of RyR2 degradation by the use of chaperone or proteasome inhibitors for cancer therapy may be involved in the cardiotoxic effects of these drugs.34
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico, FONDECYT (1110257 to P.D., Post-doctoral Fellowship 3110039 to Z.P.); Fondo de Investigación Avanzada en Áreas Prioritarias, FONDAP (15010006 to P.D., S.L.), and Anillo de Investigación de Ciencia y Tecnología ACT1111 (to S.L. and J.A.H.) from the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT), Santiago, Chile and by grants from the NIH (HL-075173, to J.A.H.; HL-080144, to J.A.H.; HL-090842, to J.A.H.), AHA (0640084N, to J.A.H.), ADA (7-08-MN-21-ADA, to J.A.H.), and the AHA-Jon Holden DeHaan Foundation (0970518N, to J.A.H.).
Acknowledgements
The authors thank Fidel Albornoz for his excellent technical assistance.
Conflict of interest: none declared.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}