





Abstract
Inflammation and angiogenesis are frequently coupled in pathological situations such as atherosclerosis, diabetes, and arthritis. The inflammatory response increases capillary permeability and induces endothelial activation, which, when persistent, results in capillary sprouting. This inflammation-induced angiogenesis and the subsequent remodelling steps are in large part mediated by extracellular matrix (ECM) proteins and proteases. The focal increase in capillary permeability is an early consequence of inflammation, and results in the deposition of a provisional fibrin matrix. Subsequently, ECM turnover by proteases permits an invasive program by specialized endothelial cells whose phenotype can be regulated by inflammatory stimuli. ECM activity also provides specific mechanical forces, exposes cryptic adhesion sites, and releases biologically active fragments (matrikines) and matrix-sequestered growth factors, all of which are critical for vascular morphogenesis. Further matrix remodelling and vascular regression contribute to the resolution of the inflammatory response and facilitate tissue repair.
1. Introduction
Mature endothelial cells (ECs) must continuously accommodate the needs of the surrounding tissue, both during normal physiology (e.g. increasing muscle blood flow during exercise) and in pathological settings (e.g. inflammation and tumourigenesis). This requires a wide range of responses that allow both acute and chronic remodelling of the vessel wall. In some cases, vascular adaptation entails physical expansion of vascular beds through an angiogenic process, which is often preceded by an inflammatory event and extensive matrix remodelling.
One of the hallmarks of inflammation, resulting from either systemic stimuli or local injury, is an increase in vascular permeability, frequently driven by an excess of vascular endothelial growth factor (VEGF), nitric oxide, or other mediators. The increased vascular permeability allows plasma components and inflammatory cells to exit the bloodstream. Two of the chief consequences of this are the formation of a provisional matrix from plasma proteins and the exit of leucocytes to the subendothelial space, initiating and sustaining the inflammatory response. The resulting acute inflammation can induce an angiogenic response, producing a highly vascularized granulation tissue, as occurs during wound healing.1 Resolution of this inflammation and the onset of lasting tissue repair cause the newly formed vasculature to regress, resulting in the restoration of homeostatic control. In the absence of vascular regression, positive feedback mechanisms operating between vessels and the inflammatory infiltrate sustain the new vasculature and further exacerbate the inflammatory response.2 This is the case in pathologies such as atherosclerosis, rheumatoid arthritis, and psoriasis,3 in which the decision between sustained inflammation or tissue repair is significantly influenced by the composition and turnover of the extracellular matrix (ECM).
The prevalent mode of vascular expansion is endothelial sprouting, which involves the invasion of avascular areas by proliferating and migrating ECs. In a nascent sprout, three phenotypically distinct EC types can be recognized: tip, stalk, and phalanx.4 Specification of tip cells is essential for capillary sprouting and is regulated by multiple inflammatory stimuli. Tip cells express the specific proteolytic machinery for migration and invasion, including membrane-type matrix metalloproteinase 1 (MT1-MMP), which plays a major role in this process.
In this review, we summarize the contributions of ECM remodelling to the distinct steps of the angiogenic response associated with inflammation (Figure 1).
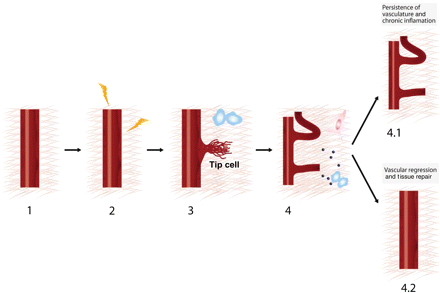
Progressive steps in inflammation-driven angiogenesis. (1) Quiescent vasculature. (2) Inflammation induced by systemic or local sources activates the angiogenic program by increasing vessel permeability and destabilizing EC junctions. (3) Proteolysis of the ECM by the endothelial tip cell during capillary sprouting induced by inflammatory stimuli. (4) ECM-driven mechanical forces, ECM-derived cryptic sites and matrikines, and growth factor signals during inflammation-mediated angiogenesis. Inflammation-induced angiogenesis can have two distinct fates: 4.1, persistence of vasculature and chronic inflammation and 4.2, vascular regression and tissue repair.
2. First steps in inflammation-induced angiogenesis: vascular permeability and EC activation
The barrier function of blood vessels is essential for homeostasis and is maintained by two types of specialized endothelial junctional complex: tight junctions and adherens junctions.5 Tight junctions are organized by claudins, occludins, and junctional adhesion molecules, whereas adherens junctions are organized by catenins and cadherins, mainly VE-cadherin.5 Together, the extracellular associations of these complexes and their intracellular links to the cytoskeleton maintain and actively regulate the vascular barrier.6 The dynamic and local control of vascular permeability enables macromolecular transport, immune surveillance, and the rapid generation of a fibrin-rich provisional matrix via the deposition of serum proteins (for reviews see Mehta and Malik7, Wu,8 Roberts and Palade9). The enhanced vascular permeability produced in response to systemic or local inflammatory stimuli increases the deposition of this provisional matrix, which provides a scaffold for the migration of infiltrating leucocytes and a template for capillary sprouting. It is probably not a coincidence that most permeability inducers are also pro-angiogenic molecules and that only a few molecules, such as fibroblast growth factor (FGF)-2, exclusively promote angiogenesis10 (Table 1).
Effect of distinct growth factors and inflammatory stimuli on vascular permeability, EC activation, and ECM proteolysis by endothelial tip cells
| Vascular permeability | EC activation (pro-angiogenic phenotype) | ECM proteolysis by endothelial tip cells | |
|---|---|---|---|
| Growth factors | |||
| VEGF | Increases permeability via adherens junctions, particularly VE-cadherin.6,98 | The main angiogenic factor described.99 Permeability and angiogenic functions have been partially dissected100 | Enriched expression in endothelial tip cells. Promotes tip cell invasiveness4 |
| Angiopoietin 1 | Blocks vascular permeability by modulating the cytoskeleton101 | Pro-angiogenic101 | No defined role |
| FGF | No reported effects on vascular permeability | An effective promoter of angiogenesis102 | Might regulate endothelial tip cell migration103 |
| Cytokines and chemokines | |||
| TNFα | Increases vascular permeability104 | Promotes angiogenesis104 | Induces an endothelial tip cell-like phenotype via the NF-κB pathway105 |
| CCL2/MCP-1 | Might indirectly increase vascular permeability106 | Promotes angiogenesis in vitro and in vivo106 | Induces MT1-MMP clustering and activity in ECs28 |
| Other bioactive molecules | |||
| Bradykinin | Increases vascular permeability in vitro and in vivo107 | Might promote angiogenesis107 | Can induce an endothelial tip cell-like phenotype108 |
| Nitric oxide | Downstream effector of bradykinin and can increase vascular permeability109 | Promotes angiogenesis109 | Induces MT1-MMP clustering and activity in ECs26 |
| PGE2 | Conflicting information | Promotes angiogenesis in vitro and in vivo110,111 | Induces MT1-MMP clustering and activity in ECs27 |
| Prostacyclin | Decreases permeability, probably by modulating stress fibres and cytoskeleton31,112 | No defined role | No defined role |
| S1P | S1P receptors can stabilize endothelial barrier integrity in vitro and in vivo via actions on the cytoskeleton113–115 | Promotes angiogenesis116 | Induces the formation of lamellipodia in ECs and cooperates with MT1-MMP during angiogenesis117,118 |
| Thrombin | Increases vascular permeability via an action on the cytoskeleton119 | Conflicting information | No defined role |
| Vascular permeability | EC activation (pro-angiogenic phenotype) | ECM proteolysis by endothelial tip cells | |
|---|---|---|---|
| Growth factors | |||
| VEGF | Increases permeability via adherens junctions, particularly VE-cadherin.6,98 | The main angiogenic factor described.99 Permeability and angiogenic functions have been partially dissected100 | Enriched expression in endothelial tip cells. Promotes tip cell invasiveness4 |
| Angiopoietin 1 | Blocks vascular permeability by modulating the cytoskeleton101 | Pro-angiogenic101 | No defined role |
| FGF | No reported effects on vascular permeability | An effective promoter of angiogenesis102 | Might regulate endothelial tip cell migration103 |
| Cytokines and chemokines | |||
| TNFα | Increases vascular permeability104 | Promotes angiogenesis104 | Induces an endothelial tip cell-like phenotype via the NF-κB pathway105 |
| CCL2/MCP-1 | Might indirectly increase vascular permeability106 | Promotes angiogenesis in vitro and in vivo106 | Induces MT1-MMP clustering and activity in ECs28 |
| Other bioactive molecules | |||
| Bradykinin | Increases vascular permeability in vitro and in vivo107 | Might promote angiogenesis107 | Can induce an endothelial tip cell-like phenotype108 |
| Nitric oxide | Downstream effector of bradykinin and can increase vascular permeability109 | Promotes angiogenesis109 | Induces MT1-MMP clustering and activity in ECs26 |
| PGE2 | Conflicting information | Promotes angiogenesis in vitro and in vivo110,111 | Induces MT1-MMP clustering and activity in ECs27 |
| Prostacyclin | Decreases permeability, probably by modulating stress fibres and cytoskeleton31,112 | No defined role | No defined role |
| S1P | S1P receptors can stabilize endothelial barrier integrity in vitro and in vivo via actions on the cytoskeleton113–115 | Promotes angiogenesis116 | Induces the formation of lamellipodia in ECs and cooperates with MT1-MMP during angiogenesis117,118 |
| Thrombin | Increases vascular permeability via an action on the cytoskeleton119 | Conflicting information | No defined role |
VEGF, vascular endothelial growth factor; S1P, sphingosine-1-phosphate.
Effect of distinct growth factors and inflammatory stimuli on vascular permeability, EC activation, and ECM proteolysis by endothelial tip cells
| Vascular permeability | EC activation (pro-angiogenic phenotype) | ECM proteolysis by endothelial tip cells | |
|---|---|---|---|
| Growth factors | |||
| VEGF | Increases permeability via adherens junctions, particularly VE-cadherin.6,98 | The main angiogenic factor described.99 Permeability and angiogenic functions have been partially dissected100 | Enriched expression in endothelial tip cells. Promotes tip cell invasiveness4 |
| Angiopoietin 1 | Blocks vascular permeability by modulating the cytoskeleton101 | Pro-angiogenic101 | No defined role |
| FGF | No reported effects on vascular permeability | An effective promoter of angiogenesis102 | Might regulate endothelial tip cell migration103 |
| Cytokines and chemokines | |||
| TNFα | Increases vascular permeability104 | Promotes angiogenesis104 | Induces an endothelial tip cell-like phenotype via the NF-κB pathway105 |
| CCL2/MCP-1 | Might indirectly increase vascular permeability106 | Promotes angiogenesis in vitro and in vivo106 | Induces MT1-MMP clustering and activity in ECs28 |
| Other bioactive molecules | |||
| Bradykinin | Increases vascular permeability in vitro and in vivo107 | Might promote angiogenesis107 | Can induce an endothelial tip cell-like phenotype108 |
| Nitric oxide | Downstream effector of bradykinin and can increase vascular permeability109 | Promotes angiogenesis109 | Induces MT1-MMP clustering and activity in ECs26 |
| PGE2 | Conflicting information | Promotes angiogenesis in vitro and in vivo110,111 | Induces MT1-MMP clustering and activity in ECs27 |
| Prostacyclin | Decreases permeability, probably by modulating stress fibres and cytoskeleton31,112 | No defined role | No defined role |
| S1P | S1P receptors can stabilize endothelial barrier integrity in vitro and in vivo via actions on the cytoskeleton113–115 | Promotes angiogenesis116 | Induces the formation of lamellipodia in ECs and cooperates with MT1-MMP during angiogenesis117,118 |
| Thrombin | Increases vascular permeability via an action on the cytoskeleton119 | Conflicting information | No defined role |
| Vascular permeability | EC activation (pro-angiogenic phenotype) | ECM proteolysis by endothelial tip cells | |
|---|---|---|---|
| Growth factors | |||
| VEGF | Increases permeability via adherens junctions, particularly VE-cadherin.6,98 | The main angiogenic factor described.99 Permeability and angiogenic functions have been partially dissected100 | Enriched expression in endothelial tip cells. Promotes tip cell invasiveness4 |
| Angiopoietin 1 | Blocks vascular permeability by modulating the cytoskeleton101 | Pro-angiogenic101 | No defined role |
| FGF | No reported effects on vascular permeability | An effective promoter of angiogenesis102 | Might regulate endothelial tip cell migration103 |
| Cytokines and chemokines | |||
| TNFα | Increases vascular permeability104 | Promotes angiogenesis104 | Induces an endothelial tip cell-like phenotype via the NF-κB pathway105 |
| CCL2/MCP-1 | Might indirectly increase vascular permeability106 | Promotes angiogenesis in vitro and in vivo106 | Induces MT1-MMP clustering and activity in ECs28 |
| Other bioactive molecules | |||
| Bradykinin | Increases vascular permeability in vitro and in vivo107 | Might promote angiogenesis107 | Can induce an endothelial tip cell-like phenotype108 |
| Nitric oxide | Downstream effector of bradykinin and can increase vascular permeability109 | Promotes angiogenesis109 | Induces MT1-MMP clustering and activity in ECs26 |
| PGE2 | Conflicting information | Promotes angiogenesis in vitro and in vivo110,111 | Induces MT1-MMP clustering and activity in ECs27 |
| Prostacyclin | Decreases permeability, probably by modulating stress fibres and cytoskeleton31,112 | No defined role | No defined role |
| S1P | S1P receptors can stabilize endothelial barrier integrity in vitro and in vivo via actions on the cytoskeleton113–115 | Promotes angiogenesis116 | Induces the formation of lamellipodia in ECs and cooperates with MT1-MMP during angiogenesis117,118 |
| Thrombin | Increases vascular permeability via an action on the cytoskeleton119 | Conflicting information | No defined role |
VEGF, vascular endothelial growth factor; S1P, sphingosine-1-phosphate.
The term endothelial activation describes both a series of molecular and cellular changes associated with the ability of ECs to recruit inflammatory cells11 and also the molecular program that leads ECs to initiate an angiogenic response. In inflammatory situations, the exit of leucocytes is associated with a temporary disruption of the vascular barrier, extravasation of plasma, and deposition of fibrin. If the inflammatory stimulus persists, more definitive and permanent changes in endothelial junctional complexes enable ECs to depart from the parental vessel and engage in the formation of a nascent sprout by invading the provisional scaffold matrix (Figure 2). These activated ECs change their cellular features and molecular expression profile and become endothelial tip cells.4 Tip cells are highly polarized and proliferate poorly and are especially suited to navigate into the surrounding tissue. In contrast, the non-migratory stalk and phalanx cells form the lumen and facilitate stabilization of the nascent vessel.4 Expression analysis of nascent sprouts and endothelial tip cells revealed the presence of VEGFR2, VEGFR3, Delta-like 4 (Dll4), angiomotin, and other molecules.12–14 These specialized cells initiate a pro-angiogenic program that includes active proteolysis and invasion of adjacent tissues.
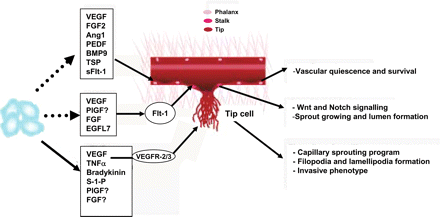
Responsiveness of distinct EC types to inflammatory and angiogenic agents. Soluble factors secreted by inflammatory, accessory, and ECs affect the fate of the different EC types in the sprout (phalanx, stalk, and tip cells), and in particular regulate the induction and specification of tip cells and therefore of capillary sprouting (adapted from De Smet et al.4).
3. Endothelial tip cells induce ECM proteolysis during inflammation-induced capillary sprouting
Capillary sprouting is one of the main mechanisms by which new vessels are formed in the adult,15 and VEGF is the most significant inducer of this response. Most of the biological functions initiated by VEGF are transduced by VEGFR2, one of the two VEGF tyrosine kinase receptors. Activation of VEGFR2 can be modulated by gradients of soluble VEGFR1 (Flt-1) that convey important local cues during capillary sprouting.16 These fine-tuning events direct and guide the tip cell.
Tip cells have specific phenotypic features including the ability to migrate in response to VEGF gradients and to invade the ECM.4 Tip cells are also provided with the enzymatic machinery for invasion through tissue barriers such as the basement membrane, interstitial matrix, and the provisional fibrin matrix deposited in response to increased vascular permeability. Members of the metzincin superfamily of proteases play important and distinct roles during capillary morphogenesis; among the proteases implicated are the matrix metalloproteinases (MMP), MT1-MMP, MT2-MMP, MT3-MMP, MMP-2, MMP-3, MMP-7, MMP-9, and MMP-13, and the ADAM family proteins, ADAM-10, ADAM-15 ADAM-17, and ADAMTS1 (reviewed by Ghajar et al.17 and van Hinsbergh and Koolwijk18). However, the functional relevance of these proteases to the invasive phenotype of endothelial tip cells remains undefined, except for the contribution of MT1-MMP.
MT1-MMP is a membrane-anchored pericellular collagenase that plays a major role in angiogenesis.19 MT1-MMP is the main endothelial fibrinolysin and collagenase responsible for EC sprouting within 3D matrices and for capillary sprouting in vivo.20,21 MT1-MMP is also required for the formation of vascular tunnels that eventually serve as 3D scaffolds for the guidance of EC, the formation of capillaries, and their subsequent stabilization.22 In the vessel, MT1-MMP expression is inhibited by pericyte–EC interactions. This restricts MT1-MMP expression to tip cells that lack mural cells.23 Thus, MT1-MMP is likely to regulate matrix remodelling at the leading edge of the developing sprout. Mathematical modelling of collagenase activities in the nascent sprout suggests that MT1-MMP, together with MMP2 and TIMP2, is one of the factors responsible for the ECM proteolysis required for invasion by endothelial tip cells during capillary sprouting.24 However, the role of MT1-MMP in the regulation of endothelial tip cells in vivo remains to be explored.
Distinct soluble factors and signalling pathways regulate the phenotype and functions of the three types of ECs (phalanx, stalk, and tip cells) (Figure 2). In addition to specification of the tip cell phenotype by VEGF gradients and Dll4/Notch signalling,4,14,25 tip cells can also be induced by inflammatory signals via alternative pathways. For example, TNFα, NF-κB, bradykinin, and sphingosine-1-phosphate can induce either the full tip cell phenotype or the formation of filopodia and lamellipodia characteristic of these migratory invading cells (Table 1). We have demonstrated that the inflammatory mediators nitric oxide (a downstream effector of bradykinin), prostaglandin E2 (PGE2), and the chemokine MCP-1/CCL2 increase the cell surface clustering and activity of MT1-MMP in human ECs and that all these factors require MT1-MMP for the induction of capillary tube formation.26–28 A more detailed analysis of the in vitro angiogenic response to a battery of growth factors and cytokines showed that MT1-MMP is widely, though not universally, required for angiogenesis.29 It will therefore be interesting to explore whether the requirement for MT1-MMP during angiogenesis mediated by these agents is linked to their putative ability to induce the endothelial tip cell phenotype. The molecular mechanisms by which the signalling pathways induced by nitric oxide, PGE2, and MCP-1/CCL2 converge to induce MT1-MMP membrane clustering and activity are poorly defined, but may involve downstream effectors such as Rac1 GTPase activity and actin polymerization26–28,30,31 (Figure 3). Notably, several stimuli that activate GTPases in endothelial tip cells also induce the formation of filopodia and lamellipodia, which are critical for the invasive function of these cells.4
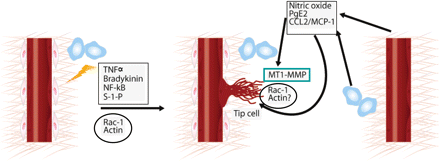
Crosstalk between inflammation, endothelial tip cells, and MT1-MMP. Tip cells can be induced by distinct inflammatory pathways (TNFα, bradykinin, NF-κB, and S-1-P) that converge on Rac1 activation and actin polymerization.4 MT1-MMP expression is restricted to the tip cells.23 Several inflammatory mediators (nitric oxide, PGE2, and CCL2/MCP-1) induce MT1-MMP clustering and activity, probably through the activation of Rac1 and actin polymerization, thereby inducing the endothelial tip cell-like phenotype.26–28
One interesting question is whether the induction of endothelial tip cells and capillary sprouting in vivo are affected by the inflammatory infiltrate and leucocyte-generated proteases. Several reports have highlighted specific contributions of inflammatory cells, particularly monocytes/macrophages, to angiogenesis. In some cases, these interactions appear to centre on endothelial tip cells. For example, in a model of hypoxia-induced retinal neovascularization, proteolytic cleavage of VEGF by macrophage-derived MMPs is required to induce the phenotype and proper guidance of endothelial tip cells.32 Monocyte/macrophages also contribute to the formation of tubular endothelial structures in Matrigel models, to ischaemia-induced lung angiogenesis in vivo and to VEGFR-3-mediated angiogenesis and lymphangiogenesis.33–35 Stromal-derived soluble MT1-MMP activates pro-MMP-2 during rat skin wound healing;36 whether active MT1-MMP is released from monocytes during inflammation37,38 and impacts the endothelial tip cell invasive program remains to be explored. In contrast to monocytes, lymphocytes limit angiogenesis during ischaemia-induced lung angiogenesis.34
Inflammatory (M1) and alternatively activated (M2) macrophages attract mesangioblasts to damaged muscle through different molecular pathways.39 Incipient angiogenic vessels can also recruit endothelial progenitors and circulating mononuclear cells; however, the mechanism by which these cells incorporate into the vascular sprout or modulate ECM remodelling have not been fully elucidated.40 Several studies have also highlighted the supportive function of monocytes in pathological neovascularization.41 In fact, elimination of Tie-2 expressing monocytes using a ‘suicide approach’ significantly impairs the angiogenic response.42 Overall, there is growing evidence for a critical crosstalk between inflammatory and ECs during vascular morphogenesis, particularly in adult settings.
4. ECM-driven signalling during angiogenesis
Although the ECM was traditionally considered the ‘glue’ that holds tissues together, it is clear now that it has broad impacts on cell behaviour and vascular morphogenesis through a wide range of mechanisms.43
4.1 ECM drives mechanical forces in inflammatory vascularization
Pathological states such as hypertension are characterized by sustained changes in pressure or flow. These changes result in dynamic artery remodelling, including changes to the shape and physical properties of the artery wall that attempt to restore homeostatic intimal shear stress and flow. This process of vessel remodelling involves ECM alterations and seems to require macrophages and MMP-9.44,45 The impact of these changes to ECM composition and structure on vascular morphogenesis is being elucidated.46–49 For example, a mathematical model of 2D sprouting has shown that matrix density influences sprout velocity and branching, whereas ECM fibre alignment correlates with EC shape and orientation.48 Moreover, degradation of matrix components at low ECM density inhibits angiogenesis, whereas matrix degradation at high ECM density promotes it. Thus, the complexity and dynamics of the ECM during capillary sprouting can influence the fate of new capillaries.
The physical properties and composition of the ECM are also critical for the generation of appropriate tensional forces. These forces can be generated by ECs, although tensional forces established by myofibroblasts seem to be more important during angiogenesis associated with the formation of granulation tissue.47,50 In fact, non-angiogenic translocation of the vasculature by myofibroblast-driven ECM tensional forces is observed during early vascular expansion in this model.50 Whether inflammatory cells contribute to these biomechanical forces in angiogenesis remains to be explored.
4.2 ECM-derived adhesion sites and active fragments in inflammation-induced angiogenesis
4.2.1 Matricryptic sites
The ultimate fate of ECs during inflammation is regulated by their interaction with ECM molecules through specific adhesion receptors that affect EC signalling and vessel remodelling.51 The dynamic nature of the ECM during the angiogenic response is partially imposed by the proteolytic processing of its components through the activity of MMPs and other proteases. Davis et al.52 coined the term matricryptic sites 10 years ago to describe ‘perturbations’ in ECM molecules, including proteolysis but also mechanical forces, novel interactions, multimerization, and conformational changes, that can expose recognition sites for EC adhesion receptors. Matricryptic sites can be found in diverse ECM proteins, and it is likely that proteases produced by macrophages contribute significantly to the generation of these sites.53,54 The critical matricryptic site Arg-Gly-Asp (RGD) is present in fibronectin and other ECM proteins such as collagens, vitronectin, and osteopontin.55 This RGD motif is exposed by proteolysis and can bind α5β1 and αvβ3/αvβ5 integrins, thus impacting EC adhesion, migration, proliferation, survival, and cell–cell interactions during angiogenesis.51 The use of soluble peptides to compete with RGD binding to integrins has therapeutic potential in disorders characterized by exacerbated or aberrant vascularization, such as cancer and chronic inflammation.56 However, recent evidence shows that low doses of RGD peptides increase angiogenesis and tumour growth by inducing VEGFR2 and β3 integrin recycling.57 These results provide an explanation for previous unsuccessful trials and highlight the need for fine-tuned regulation of the interaction of matricryptic sites with integrins during angiogenesis.
4.2.2 Matrikines
Matrikines, fragments of ECM molecules with biological functions distinct from those of the parental protein, were identified about two decades ago. They are generated by the proteolytic cleavage of ECM molecules by proteases including serine proteases and MMPs.58 As the initial report of the plasminogen-fragment angiostatin by Judah Folkman's group,59 a variety of matrikines have been identified and their role in angiogenesis explored. MMPs and matrikines, in conjunction with other pro- or anti-angiogenic factors, are likely to act in concert during any step of angiogenesis.
Matrikines are involved in wound healing,60 but the contribution of macrophage-derived MMPs53 to the production of matrikines in this and other inflammation-driven angiogenesis contexts deserves further investigation. Matrikines are most frequently derived from primary components of the basement membrane such as collagen IV (arresten, canstatin, tumstatin, and metastatin) or from the interstitial matrix surrounding the vasculature (endostatin and neostatin, derived from collagen XVIII).48 Active fragments of other ECM molecules include vastatin (collagen VIII), restin (collagen XV), anastellin (fibronectin), and endorepellin (from perlecan), all of which display anti-angiogenic activities (Table 2). The function of peptides derived from elastin, SPARC, and thrombospondins (TSPs) have also been explored in the cardiovascular system in general and in the context of inflammation-induced angiogenesis58 (Table 2). Both TSP-1 and TSP-2 exhibit inhibitory functions during wound healing angiogenesis.61 Although full-length TSPs and proteolytically derived domains display anti-angiogenic activity,58 smaller fragments are more effective.62 Most of the matrikines impact endothelial behaviour by competing with intact ECM components for interaction with integrins.51 For instance, angiostatin, tumstatin, and endostatin binds to αvβ3, angiostatin to α9β1, arresten to α1β1, and tumstatin to α5β1.58
List of selected matrikines and their function in inflammation and angiogenesis
| Matrikine | Original protein | Mechanism of action/receptor binding | Function/activity |
|---|---|---|---|
| Angiostatin | Plasminogen | αvβ3 α9β1 | Anti-angiogenic |
| Arresten | Basement membrane collagen IV | α1β1 | Anti-angiogenic |
| Canstatin | Basement membrane collagen IV | Anti-angiogenic | |
| Tumstatin | Basement membrane collagen IV | αvβ3 αα5β1 | Anti-angiogenic |
| Metastatin | Basement membrane collagen IV | Anti-angiogenic | |
| Endostatin | Collagen XVIII | αvβ3 | Anti-angiogenic |
| Neostatin | Collagen XVIII | Anti-angiogenic | |
| Vastatin | Collagen VIII | Anti-angiogenic | |
| Restin | Collagen XV | Anti-angiogenic | |
| Anastellin | Fibronectin | Anti-angiogenic | |
| Endorepellin | Perlecan | Anti-angiogenic | |
| Peptides derived from | Elastin | Pro-angiogenic | |
| Peptides derived from | SPARC | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | TSPs | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | Collagen types I and IV, elastin, fibronectin, laminins, entactin/nidogen, TSP, and hyaluronan | Elastin binding protein (67 kDa), l-selectin, Integrins, CXCR1 and CXCR2 | Chemotactic activity for inflammatory cells in vitro |
| Peptides from | Increase in vitro production of: | ||
|
| ||
| Peptides from | Promote in vivo | ||
|
|
| Matrikine | Original protein | Mechanism of action/receptor binding | Function/activity |
|---|---|---|---|
| Angiostatin | Plasminogen | αvβ3 α9β1 | Anti-angiogenic |
| Arresten | Basement membrane collagen IV | α1β1 | Anti-angiogenic |
| Canstatin | Basement membrane collagen IV | Anti-angiogenic | |
| Tumstatin | Basement membrane collagen IV | αvβ3 αα5β1 | Anti-angiogenic |
| Metastatin | Basement membrane collagen IV | Anti-angiogenic | |
| Endostatin | Collagen XVIII | αvβ3 | Anti-angiogenic |
| Neostatin | Collagen XVIII | Anti-angiogenic | |
| Vastatin | Collagen VIII | Anti-angiogenic | |
| Restin | Collagen XV | Anti-angiogenic | |
| Anastellin | Fibronectin | Anti-angiogenic | |
| Endorepellin | Perlecan | Anti-angiogenic | |
| Peptides derived from | Elastin | Pro-angiogenic | |
| Peptides derived from | SPARC | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | TSPs | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | Collagen types I and IV, elastin, fibronectin, laminins, entactin/nidogen, TSP, and hyaluronan | Elastin binding protein (67 kDa), l-selectin, Integrins, CXCR1 and CXCR2 | Chemotactic activity for inflammatory cells in vitro |
| Peptides from | Increase in vitro production of: | ||
|
| ||
| Peptides from | Promote in vivo | ||
|
|
List of selected matrikines and their function in inflammation and angiogenesis
| Matrikine | Original protein | Mechanism of action/receptor binding | Function/activity |
|---|---|---|---|
| Angiostatin | Plasminogen | αvβ3 α9β1 | Anti-angiogenic |
| Arresten | Basement membrane collagen IV | α1β1 | Anti-angiogenic |
| Canstatin | Basement membrane collagen IV | Anti-angiogenic | |
| Tumstatin | Basement membrane collagen IV | αvβ3 αα5β1 | Anti-angiogenic |
| Metastatin | Basement membrane collagen IV | Anti-angiogenic | |
| Endostatin | Collagen XVIII | αvβ3 | Anti-angiogenic |
| Neostatin | Collagen XVIII | Anti-angiogenic | |
| Vastatin | Collagen VIII | Anti-angiogenic | |
| Restin | Collagen XV | Anti-angiogenic | |
| Anastellin | Fibronectin | Anti-angiogenic | |
| Endorepellin | Perlecan | Anti-angiogenic | |
| Peptides derived from | Elastin | Pro-angiogenic | |
| Peptides derived from | SPARC | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | TSPs | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | Collagen types I and IV, elastin, fibronectin, laminins, entactin/nidogen, TSP, and hyaluronan | Elastin binding protein (67 kDa), l-selectin, Integrins, CXCR1 and CXCR2 | Chemotactic activity for inflammatory cells in vitro |
| Peptides from | Increase in vitro production of: | ||
|
| ||
| Peptides from | Promote in vivo | ||
|
|
| Matrikine | Original protein | Mechanism of action/receptor binding | Function/activity |
|---|---|---|---|
| Angiostatin | Plasminogen | αvβ3 α9β1 | Anti-angiogenic |
| Arresten | Basement membrane collagen IV | α1β1 | Anti-angiogenic |
| Canstatin | Basement membrane collagen IV | Anti-angiogenic | |
| Tumstatin | Basement membrane collagen IV | αvβ3 αα5β1 | Anti-angiogenic |
| Metastatin | Basement membrane collagen IV | Anti-angiogenic | |
| Endostatin | Collagen XVIII | αvβ3 | Anti-angiogenic |
| Neostatin | Collagen XVIII | Anti-angiogenic | |
| Vastatin | Collagen VIII | Anti-angiogenic | |
| Restin | Collagen XV | Anti-angiogenic | |
| Anastellin | Fibronectin | Anti-angiogenic | |
| Endorepellin | Perlecan | Anti-angiogenic | |
| Peptides derived from | Elastin | Pro-angiogenic | |
| Peptides derived from | SPARC | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | TSPs | Pro-angiogenic/anti-angiogenic | |
| Peptides derived from | Collagen types I and IV, elastin, fibronectin, laminins, entactin/nidogen, TSP, and hyaluronan | Elastin binding protein (67 kDa), l-selectin, Integrins, CXCR1 and CXCR2 | Chemotactic activity for inflammatory cells in vitro |
| Peptides from | Increase in vitro production of: | ||
|
| ||
| Peptides from | Promote in vivo | ||
|
|
It is also becoming clear that proteolysis-derived ECM fragments affect the inflammatory response (for a review see Adair-Kirk and Senior63 and Table 2). Fragments of collagen types I and IV, elastin, fibronectin, laminins, entactin/nidogen, TSP, and hyaluronan display chemotactic activity for inflammatory cells in vitro. These fragments can also induce specific gene expression programs in inflammatory cells, in particular the expression of proteases and cytokines that may eventually impact capillary sprouting and angiogenesis. Peptides fragments of fibronectin and laminin, for example, increase monocyte/macrophage expression of a variety of proteases including MT1-MMP.64,65 Notably, laminin, elastin, and collagen fragments can also induce protease and cytokine production and leucocyte recruitment in vivo63,66 (Table 2).
4.3 ECM modulates growth factors that regulate vascular morphogenesis, remodelling, and responses to inflammatory stimuli
4.3.1 VEGFs and ECM
Like many other growth factors, VEGF binds to ECM molecules. This interaction is regulated by alternative splicing, which generates a variety of VEGF isoforms with distinct affinities for specific ECM proteins. Alternatively, VEGF–ECM interaction can also be altered post-translationally by proteolytic processing (Figure 4). In this case, the length of the C-terminal region can be modified depending on the availability of active proteases. Specifically, plasmin and a subset of MMPs can cleave the C-terminal region of VEGF to release the bioactive growth factor (receptor-binding domain) from its anchorage site in the ECM.67
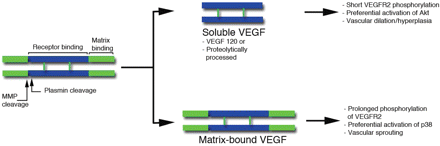
Schematic representation of VEGF. Two monomers are held together in an anti-parallel orientation by disulfide bonds. The central region (blue) is the receptor binding domain, which binds both VEGFR1 and 2, and is encoded by exons 2–5. The C-terminal region (green) includes the matrix-binding motif (encoded by a variable number and combination of exons 6a, 6b, and 7). Amino acids encoded by exon 8 are present in all VEGF forms. Plasmin and some matrix metalloproteases can sever (by proteolysis) the receptor-binding motif from the extracellular matrix binding domain. Soluble VEGF lacks the matrix-binding region either because it is secreted as a short alternative spliced form (VEGF120) or because is modified post-translationally by plasmin or MMPs. The effects of soluble and bound forms are indicated on the right and based on our previous data.67,69
VEGF can be cleaved by plasmin at 110–111 aa and by MMPs at 113 aa. A large cohort of MMPs can process VEGF, including MMP-3, MMP-7, MMP-9 (in the presence of heparin), and MMP-19.67 These endoproteolytic events can modulate the levels of soluble and matrix-bound VEGF in tissues and thereby result in either enlarged vessels or sprouting angiogenesis (Figure 4). In this way, the number of inflammatory cells can modulate the binding of VEGF to the ECM, thereby regulating its growth factor activity.
The release of VEGF from matrix stores has been implicated in the angiogenic switch, facilitating the transition of tumours from the hyperplastic to the malignant state.68 The ability of proteases to modulate VEGF release from the ECM is thus significant in the context of specific tissues and pathological settings. All VEGF isoforms (including VEGF121) can be cleaved by plasmin (110 aa) or MMPs (113 aa). As cleaved and bound VEGF can both phosphorylate the two main tyrosine receptors, the significance of these proteolytic processing events has not been clear. Nonetheless, recent findings indicate that matrix-bound or soluble VEGF provides important contextual differences in signalling; for example, matrix anchorage leads to clustering of VEGFR2 and increases receptor internalization and downstream phosphorylation kinetics, all of which contribute to distinct endothelial responses.69
In fact, these differences between soluble and matrix-bound VEGF result in alternative modes of vascular expansion. Using engineered forms of VEGF, we found that a version mimicking cleaved soluble VEGF113 produced vascular beds with low vascular density and poor branching. In contrast, a matrix-bound VEGF mutant, resistant to cleavage by either plasmin or MMPs, induced highly branched and thin vessels that greatly facilitated tissue perfusion and tumour growth.67 Thus, soluble VEGF induces vascular hyperplasia, while sprouting angiogenesis is induced by matrix-bound VEGF.
4.3.2 FGF and ECM
The interaction of FGF with extracellular matrix proteins, particularly heparan sulfate proteoglycans, has been explored both in vitro and in vivo in elegant genetic mouse models.70–72 It is now well accepted that these interactions depend on structures determined by the sulfation pattern and by which hydroxyl group is oxidized in uronic acid residues.73 Moreover, FGFR1 levels in pancreatic beta cells are reduced by the binding of alpha6 integrin to laminin, with consequent reduction in ERK activation and proliferative activities;74 thus alterations in integrin profiles at the cell surface by matrix substrates can further contribute to the outcome of FGFR activation. Taken together, the data convincingly demonstrate that the ECM contributes significantly to finetune the complex FGF signalling network during distinct phases of embryonic development and in pathological settings in adults.
4.3.3 Transforming growth factor-beta and ECM
The interaction of Transforming growth factor (TGF)-beta with ECM not only provides a means of anchoring the ligand to the extracellular environment; it can also contribute to its activation. For example, association with thrombospodin1 mediates a physiologically relevant activation by TGF-beta.75 Interestingly, however, TSP-1 inhibits VEGF.76 Thus, it is not the identity of the matrix component but the combined association that determines whether the growth factor is activated or inhibited.
4.3.4 Platelet-derived growth factor and ECM
Platelet-derived growth factor (PDGF) also binds to matrix proteins. Using plasmon resonance assays, Gohring et al.77 identified several collagen types, laminin 1, nidogen, perlecan, and BM-40 as PDGF binding partners. Interaction with matrix proteins does not affect binding of PDGF to its receptors and suggests that matrix binding might provide storage sites for PDGF in the extracellular environment for later release upon partial matrix proteolysis, thereby regulating the association of mural cells with the growing endothelial sprout.
5. ECM remodelling during resolution of inflammation: regression of provisional vasculature
During the resolution of the inflammatory response, the provisional vasculature must regress in order to facilitate proper tissue repair.1,78 Fine-tuned adjustments to the proteolytic machinery of ECs are required for removal of the recently formed blood vessels, the provisional ECM and the leucocyte infiltrate.
One of the best physiological examples of capillary regression is the remodelling of the hyaloid vasculature in the developing retina.79–81 Although the precise mechanism of hyaloid vessel regression is not fully understood, the first step involves apoptosis of pericytes, and is followed by regression of the endothelial tubes. Macrophages are found in close association with hyaloid vessels even after these vessels became vestigial and disappear,82 suggesting that macrophages participate in the pericyte apoptosis and contribute to capillary regression.83 This was confirmed by the examination of PU.1-deficient mice, which lack macrophages. Persistence of hyaloid vasculature postnatally in these mice shows that macrophages are essential for the process.84 Specific genes required for vascular removal have been identified. For example, the Norrie disease gene product is necessary for hyaloid vessel regression; however, the mechanism is unclear, because the absence of this gene is not associated with changes in macrophages or endostatin.85 More recently, it has been shown that ninjurin1 (nerve injury-induced protein-1) is implicated in macrophage activation and interaction with vascular ECs. Neutralization of this pathway results in delayed hyaloid regression, suggesting that macrophage expression of Ninj1 amplifies the death signal through cell–cell interactions.86
Capillary regression is also evident in wound healing, during which the highly vascularized granulation tissue needs to be removed to allow proper skin repair. After capillary sprouting, the newly formed vasculature is first stabilized by the inhibition of endothelial MT1-MMP, achieved by the secretion of TIMP3 by directly adjacent vascular pericytes.87 Other MMPs induced during the formation of the granulation tissue88 contribute to vascular regression. For example, work by Davis’ group with in vitro models has identified MMP-1 and MMP-10 as proteases essential for proper capillary tube regression.88–90 Plasminogen and plasma kallikrein may also accelerate the process. The role of all these proteases in vascular regression is complex and probably includes direct degradation of ECM components and alterations to the mechanical forces that prevent the collapse of endothelial tubes. Another likely contributing factor is the leucocyte infiltrate. However, skin wound repair was normal in PU.1 null ‘macrophageless’ mice, with only a slightly enhanced early angiogenic response.91 Although this study did not specifically address the vascular regression phase, this finding suggests that there might be alternative mechanisms of vessel regression in distinct tissues, some of them independent of leucocyte infiltrate. The contribution of macrophages to the impaired regression of new vessels associated with chronic inflammatory disorders, such as the atheromatous plaque, remains unexplored.
6. Future directions
Much has been learned about the role of ECM remodelling in inflammation-driven angiogenesis and in the vascular regression necessary for proper tissue repair during pathological states. However, the molecular regulatory events that mediate tissue-specific ECM changes and the resolution of the inflammatory response are yet to be elucidated. The systems biology technologies emerging during the post-genomic era are likely to refine our knowledge of the complexity of proteins and molecular pathways involved in this process. One potential area of interest is the degradome, the battery of proteases and inhibitors expressed by a cell type under specific conditions or the protease substrate repertoire of a specific cell type. Characterization of the degradome in endothelial, mural, and accessory cells activated in distinct inflammatory contexts promises to identify new targets for the inhibition or promotion of angiogenesis. In addition, miRNAs have emerged as critical modulators of gene expression in a variety of processes including angiogenesis, ECM production, and remodelling.92–94 Alteration of miRNA activity in vivo in animal models offers a promising avenue for the development of new treatments for disorders involving ECM remodelling during pathological angiogenesis.95
Furthermore, normalization of the aberrant vasculature, rather than inhibition of its formation, may offer a better prospect for treating inflammatory diseases characterized by uncontrolled vascularization. Various targets for promoting normalization are being explored in animal models and will soon progress to clinical trials.96,97
ECM remodelling affects angiogenesis in several ways, but its contribution to vascular regression is of fundamental importance to tissue repair. The persistence of newly formed vasculature in an inflammatory context leads to the maintenance of the disorder. More studies are therefore needed to define the role of macrophages and other immune cells during the regression of neovasculature in diseases such as atherosclerosis, and may lead to strategies for genetic or pharmacological manipulation of macrophage activity to promote vessel regression and contribute to tissue repair.
Funding
This work was supported by the Spanish Ministry of Science and Innovation [SAF2008-02104 to A.G.A]; the Spanish Fondo de Investigación Sanitaria [RD06/0014/1016 to A.G.A]; the Fundación Ramón Areces [to A.G.A]; and by National Institutes of Health [RO1CA126935 to M.L.I.-A.]. The CNIC is supported by the Spanish Ministry of Science and Innovation and the Pro-CNIC Foundation.
Acknowledgements
We thank Benny Gee for graphic work and Simon Bartlett for English editing.
Conflict of interest: none declared.
References
Author notes
This article is part of the Spotlight Issue on: Mechanisms of Vascular Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}