





Abstract
Phosphatase inhibitor-1 (I-1) is a conditional amplifier of β-adrenergic signalling downstream of protein kinase A by inhibiting type-1 phosphatases only in its PKA-phosphorylated form. I-1 is downregulated in failing hearts and thus contributes to β-adrenergic desensitization. It is unclear whether this should be viewed as a predominantly adverse or protective response.
We generated transgenic mice with cardiac-specific I-1 overexpression (I-1-TG) and evaluated cardiac function and responses to catecholamines in mice with targeted disruption of the I-1 gene (I-1-KO). Both groups were compared with their wild-type (WT) littermates. I-1-TG developed cardiac hypertrophy and mild dysfunction which was accompanied by a substantial compensatory increase in PP1 abundance and activity, confounding cause–effect relationships. I-1-KO had normal heart structure with mildly reduced sensitivity, but unchanged maximal contractile responses to β-adrenergic stimulation, both in vitro and in vivo. Notably, I-1-KO were partially protected from lethal catecholamine-induced arrhythmias and from hypertrophy and dilation induced by a 7 day infusion with the β-adrenergic agonist isoprenaline. Moreover, I-1-KO exhibited a partially preserved acute β-adrenergic response after chronic isoprenaline, which was completely absent in similarly treated WT. At the molecular level, I-1-KO showed lower steady-state phosphorylation of the cardiac ryanodine receptor/Ca2+ release channel and the sarcoplasmic reticulum (SR) Ca2+-ATPase-regulating protein phospholamban. These alterations may lower the propensity for diastolic Ca2+ release and Ca2+ uptake and thus stabilize the SR and account for the protection.
Taken together, loss of I-1 attenuates detrimental effects of catecholamines on the heart, suggesting I-1 downregulation in heart failure as a beneficial desensitization mechanism and I-1 inhibition as a potential novel strategy for heart failure treatment.
1. Introduction
Chronic heart failure is characterized by a decrease in cardiac output and a compensatory activation of the sympathetic nervous system. Sympathetic overstimulation leads to desensitization and attenuated β-adrenergic responsiveness.1,2 On the one hand, β-adrenergic desensitization aggravates exercise intolerance by reducing the contractile reserve of the heart and may therefore be viewed as maladaptive. On the other hand, it could be a beneficial mechanism that protects the heart from chronic β-adrenergic overstimulation, including arrhythmias and hypertrophy.2 The development of novel therapeutic strategies fundamentally depends on these interpretations, aiming at either inhibiting or restoring the β-adrenergic sensitivity.
Several mouse models have addressed this issue. Transgenic heart-specific overexpression of β1-adrenoceptors, Gαs, PKA, or targeted deletion of phosphodiesterase 4D3 caused cardiomyopathy and/or arrhythmia and premature death.3–6 These data favour the view that sustained activation of the β-adrenergic pathway has predominantly detrimental consequences. The clinical success of β-blockers supports this conclusion. Importantly, part of the adverse consequences of β-adrenergic overstimulation appears to be mediated by increased phosphorylation of ryanodine receptors (RyR2) resulting in an increased propensity of spontaneous Ca2+ release.7–9 On the other hand, overexpression of type 6 adenylyl cyclase10 or genetic ablation of phospholamban (PLB) causing accelerated diastolic Ca2+-reuptake into the sarcoplasmic reticulum (SR) did not cause heart failure, but improved long-term function in failing hearts.11 At present, it is not clear whether these conceptually divergent findings indicate different roles of the various components of the β-adrenergic signalling pathway, distinct cellular compartments, or quantitative differences in the expression level.
A relatively newly identified element of the β-adrenergic signalling pathway in the heart is the protein phosphatase-1 inhibitor-1 (I-1). I-1 is an ubiquitously expressed cytosolic acid- and heat-stable protein,12 which becomes active upon phosphorylation at Thr35 by the cAMP-dependent PKA, whereas phosphorylation at Ser67 or Thr75 by PKCα attenuates I-1's inhibitory activity.13,14 Activation of I-1 results in specific inhibition of the catalytic subunit of protein phosphatase type-1 (PP1c), the major isoform of Ser–Thr–protein phosphatases in the heart, resulting in enhanced phosphorylation of I-1/PP1-sensitive regulatory phosphoproteins. I-1 is downregulated in human and experimental heart failure.15–18 In adult rat and in failing human cardiac myocytes, the overexpression of either wild-type (WT) I-1 or a truncated, constitutively active I-1 mutant protein enhanced β-adrenergic-dependent contractile responses and steady-state phosphorylation of PLB, emphasizing its amplifier role in this system.15,17 Moreover, transgenic or adenoviral overexpression of the truncated I-1 protected mice from contractile dysfunction in a pressure overload model. These findings collectively indicate that I-1 could be a promising therapeutic target in heart failure.19
In this study, we compared myocardial performance of I-1-overexpressing mice (I-1-TG) and knock-out mice (I-1-KO20) under basal conditions and after chronic β-adrenergic overstimulation to simulate conditions characteristic for heart failure. The main conclusions are the overexpression of full-length I-1 causes adverse compensatory upregulation of PP1c, whereas the ablation of I-1 protects against acute and chronic catecholamine stimulation without affecting heart rate.
2. Methods
2.1 Mice and isoprenaline administration
The transgenic mice were generated in the transgenic mouse facility of the University Medical Center Hamburg-Eppendorf by pro-nuclear injection of a ∼6 kb cDNA fragment consisting of the α-myosin heavy chain promoter, the rat full-length I-1 cDNA, and the simian virus 40 polyadenylation site. I-1-deficient mice (C57BL/6J) were a kind gift of Dr P. Greengard.20 Genotyping and primer details are provided in the Supplementary material online. Isoprenaline (Sigma, Deisenhofen, Germany) was delivered to mice by subcutaneously implanted osmotic minipumps (Alzet, model 1007d, Sulzfeld, Germany) that released isoprenaline in 0.9% NaCl for 7 days at a dose of 15 mg/kg per day. Control pumps delivered 0.9% NaCl solution alone. Anaesthesia was performed with ketamine (120 mg/kg) and xylazine (16 mg/kg) or isoflurane (1.5 vol%). The pumps were immersed in 0.9% NaCl overnight to achieve the full pumping rate from the start. Only littermate male mice of 12–18 weeks of age were used in this protocol. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and were approved by the governmental review board in Hamburg (G 21/1-46/04).
2.2 Echocardiographic studies
Animals were kept under temperature- and ECG-controlled anaesthesia (isoflurane, 1.5 vol%). Two-dimensional echocardiography images were obtained in a modified parasternal long- and a short-axis view at mid-papillary muscle level at a frame rate of 60 Hz (VisualSonics Vevo 660, Toronto, Canada). Echocardiographic studies were performed in unstressed mice and after intraperitoneal injection of a maximally effective dose of the β-agonist dobutamine (20 µg/g). Further details are provided in the Supplementary material online.
2.3 Left atrial force measurement
Experiments were performed at 37°C in organ baths as described previously.21 Muscle length was adjusted to maximal twitch force (Lmax). Twitch contraction was generated by electrical field stimulation at 1 Hz with 20 mA stimulation current and 5 ms pulse duration. At steady-state isometric concentration, responses to isoprenaline (0.1–10 µM) were recorded.
2.4 Cardiac catheter and pressure–volume measurements
In vivo haemodynamic analysis was performed with a four-electrode pressure–volume catheter (model SPR-839, Millar Instruments, Houston, Texas, USA) placed through the right carotid artery in the closed-chest, isoflurane-anaesthetized mouse and positioned along the longitudinal axis of the left ventricle to record chamber volume by impedance and pressure by micromanometry. Further details are provided in the Supplementary material online.
2.5 Langendorff perfusion
Hearts were subjected to retrograde perfusion at room temperature in continuously oxygenated (95% O2 + 5% CO2) modified Tyrode’s solution containing 119.8 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.05 mM MgCl2, 0.42 mM NaH2PO4, 22.6 mM NaHCO3, 0.05 mM Na2EDTA, 0.5 mM ascorbic acid, 10 mM glucose, and 5 mM pyruvate using a standard Langendorff system.
2.6 Histological analysis
Cross-sections from the mid-papillary level of the heart (∼2 mm thick) were fixed with 4% paraformaldehyde, dehydrated with graded concentrations of isopropyl-alcohol, embedded in paraffin, and cut into 4 µm slices. Sections were stained with haematoxylin/eosin. For morphometric analyses, photographs of 20 ventricular sections from WT and I-1-KO mice were taken at ×320 magnification (Zeiss IM-35, Jena, Germany). Myocyte cross-sectional areas were determined in digitized images. Only nucleated myocytes from areas with transversely cut muscle fibres were evaluated. Sirius red staining was used to detect fibrosis.
2.7 Western blot analysis, real-time reverse transcriptase—polymerase chain reaction, protein phosphatase assay, and radioligand-binding experiments
Western blotting was performed as described previously.15 Details on primary antibodies are provided in the Supplementary material online. Real-time reverse transcriptase–polymerase chain reaction (RT–PCR) was performed with primers and probes specific for I-1 and ANP with the TaqMan system (ABIPrism 7900, Applied Biosystems, Weiterstadt, Germany). I-1 protein was enriched by an optimized trichloroacetic acid extraction procedure as described before.16
Protein phosphatase activity was determined as described previously, with [32P]phosphorylase a (Sigma, Deisenhofen, Germany) as substrate, and okadaic acid (3 nM) was used to differentiate between PP1 and PP2A activities.15 Details for radioligand-binding assay is provided in the Supplementary material online.
2.8 Statistical analysis
Unpaired Student's t-test (two-sided) was used to compare means between two independent groups/samples. Paired t-test (two-sided) was used to compare values in mice before and after treatment. Dose/concentration–response curves were analysed by repeated-measures ANOVA. Survival analysis was performed by the Kaplan–Meier method, and between-group differences in survival were tested by the log-rank test. Average data are presented as mean ± SEM. Differences were considered significant when P < 0.05.
3. Results
3.1 Inhibitor-1 overexpression induces upregulation of phosphatase-1 and cardiac hypertrophy and dysfunction
To determine the long-term in vivo effects of cardiac I-1 overexpression, we generated a transgenic mouse line with cardiomyocyte-specific overexpression (Figure 1A). We identified marked I-1 overexpression in these mice by immunodetection with a custom-made antibody as described previously15 and intact phosphorylation at Thr35 (PKA-site) and Ser67 (PKC-site) using phosphorylation-specific antibodies22 (Figure 1B, left panel). In order to calculate the absolute level of I-1 in WT murine heart tissue and in I-1-TG, dilutions of recombinant rat I-1 (0.1–0.0025 µg), I-1-enriched acid extract from WT hearts (5–20 µg) and standard homogenates from I-1-TG hearts were analysed by western blotting (3 and 6 µg). These experiments revealed I-1 levels in murine WT hearts at ∼450 fmol/mg protein and ∼90 pmol/mg protein in I-1 TG hearts, indicating a ∼200-fold I-1 overexpression in I-1-TG (Figure 1B, right panel). Heart-to-body weight (HW/BW) ratio was increased by 14% in I-1-TG at 3 months of age (P < 0.05, Figure 1C) when compared with WT littermates and increased further to 20% at 10 months of age (P < 0.05, not shown), indicating spontaneous development of cardiac hypertrophy. Echocardiographic examination of cardiac function in vivo under light isoflurane anaesthesia (1.5%) revealed normal heart rate (448 ± 8 vs. 458 ± 7 b.p.m. in WT, n ≥ 15), slightly lower contractile function [fractional area shortening (FAS)] under basal conditions (P < 0.05), but a normal increase in FAS when stimulated with a half-maximal dose of dobutamine (3 µg/g i.p.; 15 vs. 13% in WT, Figure 1D). PLB phosphorylation, a known downstream target of I-1, was analysed in quickly excised hearts by western blotting and it did not differ between I-1 TG and WT littermates, neither at Ser16 nor at Thr17 (Figure 1E).
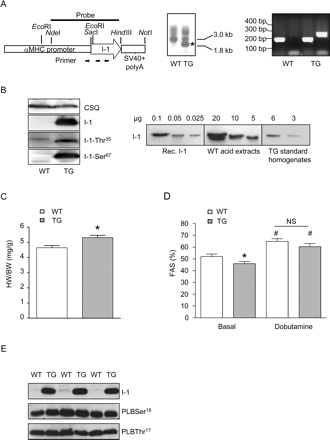
Inhibitor-1-overexpressing transgenic mice exhibit cardiac hypertrophy and slight contractile dysfunction. (A) Vector used to generate I-1 transgenic mice (TG, left panel). Southern blot (middle) and polymerase chain reaction (right panel) identifying founder (B) Western blot demonstrating the overexpression of I-1 with intact Thr-35 and Ser-67 phosphorylation sites (left panel). Dilutions of recombinant rat I-1, acid extracts from wild-type (WT) hearts (∼500-fold concentrated), and standard homogenates from transgenic mice hearts showing that I-1 is overexpressed ∼200 folds (right panel). (C) Heart weight (HW)-to-body weight (BW) ratio in wild-type and transgenic (n = 13 each) mice at 3 months of age, *P < 0.05 vs. wild type. (D) Echocardiographical assessment of fractional area shortening (FAS, n ≥ 7 each) before and after injection of a half-maximal dose of dobutamine (3 µg/g i.p.), *P < 0.05 vs. wild type, #P < 0.05 vs. before dobutamine. (E) Blots for I-1 and phospholamban (PLB) phosphorylation levels at Ser16 and Thr17 in vivo under basal conditions.
To evaluate consequences of I-1 overexpression in the absence of potentially confounding in vivo factors, we assessed PLB phosphorylation in I-1-TG ex vivo in Langendorff heart preparations. Surprisingly, PLB phosphorylation levels in TG were ∼50% lower under basal conditions, but three-fold higher than in WT after isoprenaline stimulation (1 µM, Figure 2A). The latter finding is compatible with the amplifier role of I-1. The lower basal PLB phosphorylation in I-1-TG was unexpected and pointed to compensatory mechanisms. Indeed, PP1c protein levels were approximately three-fold higher (Figure 2B) and, despite the marked overexpression of I-1, were accompanied by approximately three-fold increase in PP1c activity in crude homogenates (Figure 2C), with no significant change in PP2A activity (4.0 ± 0.5 vs. 2.5 ± 0.8 pmol/mg/min in WT, n ≥ 5). The higher PP1c protein levels were confirmed in a second independent I-1-TG line (Figure 2B) with similar I-1 overexpression levels (∼250-fold, not shown) and reflects similar observations in other PP1c regulatory/targeting subunit overexpressor lines such as inhibitor-223,24 and RGL/GM25 transgenics. These data indicate, on the one hand, functional activity of overexpressed I-1, but, on the other hand, also compensation against the unphysiologically high and unregulated I-1 levels. In support of the latter, we observed an unusual, spotted subsarcolemmal distribution of I-1 in I-1-TG instead of the more homogenous cytosolic localization recently observed by adenoviral I-1 overexpression in cardiac myocytes (Figure 2D).15,26
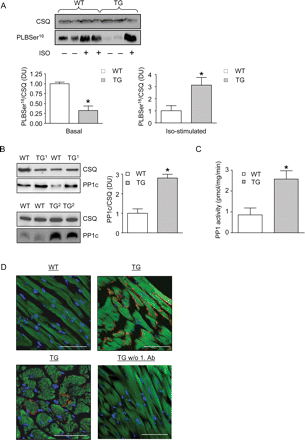
Inhibitor-1 overexpression leads to a concomitant increase of PP1c protein levels/activity and a spotted subsarcolemmal distribution of the overexpressed I-1 transgene in the cardiomyocytes. (A) Blots of samples from Langendorff-perfused wild-type (WT) and transgenic (TG) hearts treated with (n = 4 each) or without (n = 4 each) isoprenaline (10−6 M Iso) for CSQ and PLBSer16 phosphorylation, *P < 0.05 vs. wild type. (B) Blots from samples of wild type and two different transgenic lines for CSQ and phosphatase-1 (PP1c) (n ≥ 4 each), *P < 0.05 vs. wild type. (C) PP1c activity determined in samples from Langendorff-perfused hearts (n ≥ 5 each), *P < 0.05 vs. wild type. (D) Longitudinal and cross-sections from paraffin-embedded hearts were immunostained for I-1 (red), DAPI (nuclear stain, blue), and α-sarcomeric actin (green). Control staining was performed without primary antibody. Images were acquired with the same microscope settings. The bars indicate 50 µm.
3.2 Inhibitor-1-deficient mice show mild β-adrenergic desensitization, but normal basal cardiac function and normal contractile reserve
The absence of I-1 gene products in I-1-KO mice was confirmed by PCR and western blot analyses (Figure 3A). The functional consequences of I-1 deficiency were characterized initially in isolated left atria by isometric contraction experiments. The force of contraction did not differ between I-1-KO and their WT littermates, neither under basal conditions (0.8 ± 0.1 mN in I-1-KO vs. 0.9 ± 0.1 mN in WT, n = 12) nor in the presence of a maximally effective concentration of isoprenaline (10 µM; 3.8 ± 0.4 vs. 4.0 ± 0.5 mN in WT, n = 12). However, the β-agonist isoprenaline was less potent in I-1-KO than in WT (EC50 41 vs. 24 nM, P < 0.05, Figure 3B), indicating that loss of I-1 induces mild β-adrenergic desensitization.
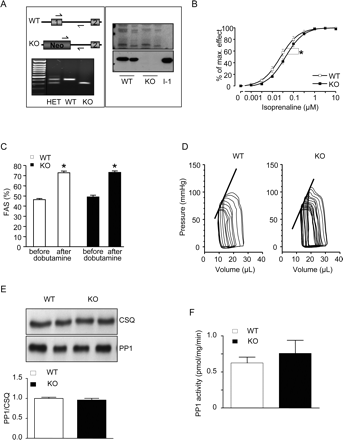
Inhibitor-1-deficient mice (I-1-KO) exhibit β-adrenergic desensitization and a normal contractile reserve. (A) Genotyping strategy of I-1-KO mice and representative polymerase chain reaction with DNA from wild-type (WT), heterozygous (HET) and homozygous mice (left panel). Immunoblot (right panel) of boiled acid extracts from skeletal muscle of wild type and I-1-KO mice (10 and 20 µg each) probed with I-1 antibodies. (B) Inotropic effect of increasing isoprenaline concentrations on isolated left atria from I-1-KO (n = 12) and wild-type littermates (wild type, n = 12) recorded in organ baths, *P < 0.05 vs. wild type. (C) Assessment of fractional area shortening (FAS) in wild type (n = 21) and KO (n = 15) before and after injection of maximal dobutamine (20 µg/g i.p.), *P < 0.05 vs. before dobutamine. (D) Representative occlusion analysis of pressure–volume loops. (E) Blots from samples of wild type (n = 5) and I-1-KO (n = 5) for CSQ and PP1c. (F) PP1c activity determined in crude homogenates with phosphorylase a as substrate in wild type (n = 5) and I-1-KO (n = 5).
I-1-KO followed up to 1 year of age presented a normal HW/BW ratio (4.8 ± 0.2 vs. 4.7 ± 0.2 mg/g in WT, n = 16). At 3 months of age, echocardiographic examination of cardiac function revealed normal heart rate (499 ± 17 vs. 485 ± 10 b.p.m. in WT, n ≥ 16), normal basal contractile parameters, and a preserved contractile response to a maximally effective dose of dobutamine (20 µg/g i.p.) in I-1-KO (Figure 3C). These results were confirmed by cardiac catheterizations and pressure–volume loop measurements in an independent cohort of animals showing identical contractile parameters in I-1-KO and WT (Supplementary material online, Table S1). Importantly, and in contrast to I-1-TG, there was no compensatory change in PP1c protein levels in I-1-KO (Figure 3E). Moreover, radioligand-binding experiments revealed no change in β-adrenoceptor density between I-1-KO and WT (15.6 ± 0.6 vs. 16.2 ± 1.4 fmol/mg in WT, n ≥ 5). Consistent with a previous report,17 PP1c activity determined in crude homogenates under basal conditions was not significantly different between I-1 KO and WT (Figure 3F).
3.3 Inhibitor-1-deficient mice are partially protected from acute isoprenaline-induced lethality under ketamine/xylazine anaesthesia
The mild phenotype in I-1-KO led us to hypothesize that relevant consequences of I-1 deficiency may become evident under conditions of higher neurohumoral stress, as it occurs in heart failure. Therefore, we implanted isoprenaline-containing minipumps under ketamine/xylazine anaesthesia to infuse isoprenaline for 7 days. Surprisingly, we observed peri-procedural death in all WT animals (n = 6; Figure 4A). Importantly, none of the mice that got the NaCl pump died. ECG analysis showed that isoprenaline-treated animals died from ventricular arrhythmias (premature ventricular beats, ventricular tachycardia, ventricular fibrillation, and bradycardia; Figure 4B). In contrast to WT, three out of seven I-1-KO survived the same procedure (Figure 4A).
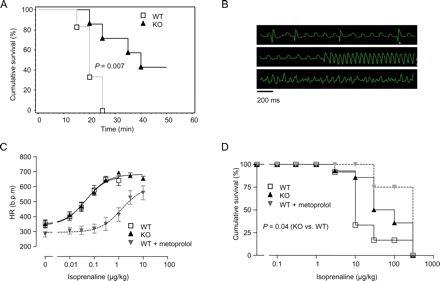
Inhibitor-1-deficient mice (I-1-KO) exhibit less susceptibility to isoprenaline-induced deaths. (A) Survival after subcutaneous implantation of isoprenaline-delivering minipumps during ketamine/xylazine anaesthesia in wild-type (WT) (n = 6) and I-1-KO (n = 7). (B) Representative ECGs recorded from a wild-type animal in 5–10 min intervals after isoprenaline pump implantation. (C and D) Chronotropic and lethal effects of increasing isoprenaline doses (0.01–300 µg/kg i.p.) in I-1-KO (n = 14), wild-type (n = 12), and wild type pre-treated with the β-blocker metoprolol (1650 µg/kg, n = 4).
In order to systematically evaluate this finding, WT, I-1-KO, and, for positive control, WT mice pre-treated with a maximally effective dose of the β-blocker metoprolol were anaesthesized with ketamine/xylazine and then subjected to increasing doses of isoprenaline injected i.p. under continuous ECG recording. As expected, metoprolol-pre-treated WT mice tolerated 25-fold higher doses of isoprenaline to increase heart rate (Figure 4C) and induce lethal arrhythmias (Figure 4D). This indicated effective β-blockade and pointed to a β1-adrenoceptor-mediated mechanism of arrhythmia. Yet, I-1-KO mice were also partially protected against isoprenaline-induced lethal arrhythmia and tolerated approximately eight-fold higher doses than WT (Figure 4D). Intriguingly, in contrast to the β-blocker-pre-treated animals, I-1-KO showed a completely normal heart rate response (Figure 4C).
3.4 Inhibitor-1-deficient mice are partially protected from chronic isoprenaline-induced hypertrophy, dilatation, and β-adrenergic desensitization
Isoflurane anaesthesia allowed the application of isoprenaline without excess mortality and was therefore used for all further experiments. In WT littermate mice, a 7 day infusion of isoprenaline induced cardiac hypertrophy as indicated by an increased HW/BW ratio (17 ± 2%, P < 0.05, Figure 5A and Supplementary material online, Table S2) and echocardiographically determined left ventricular mass (LVM: 41 ± 4%, P < 0.05, Figure 5B). These effects were less pronounced in I-1-KO (HW/BW: 9 ± 2% and LVM: 23 ± 5%; Figure 5A and B). Moreover, WT, but not I-1-KO, showed significant left ventricular dilation (LVEDD 11 ± 2%, P < 0.05, Figure 5C). Measurement of cardiomyocyte size in cross-sections confirmed less hypertrophy in isoprenaline-treated I-1-KO mice (cardiomyocyte cross-sectional area: 19 ± 2 vs. 32 ± 2% in WT, Figure 5D). In accordance with a previous report, the extent of sirius-red-stained fibrosis after isoprenaline was low in the C57Bl/6 mouse strain.27 However, interstitial fibrosis appeared to be less frequent and less extensive in I-1-KO mice (Figure 5E). In addition, quantitative PCR measurements in WT hearts revealed a greater than two-fold increase in transcript levels of the cardiac foetal gene ANP after chronic isoprenaline, which was almost absent in I-1-KO hearts (Figure 5F).
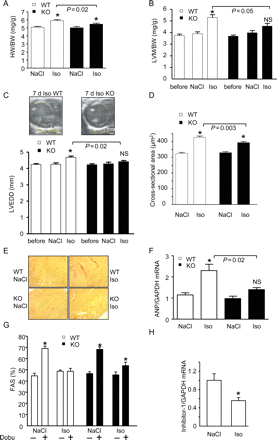
Attenuated morphological and functional deterioration following chronic isoprenaline in I-1-KO mice. (A) Heart-to-body weight ratio (HW/BW) in wild-type (WT) and I-1-KO mice treated for 7 days with NaCl or isoprenaline (n > 12 for each group). (B) Echocardiographically determined left ventricular mass normalized to body weight (LVM/BW, n ≥ 8 for each group) before implantation of the pumps (‘before’) and after 7 days of NaCl or isoprenaline, *P < 0.05 vs. NaCl; NS, not significant vs. NaCl. (C) Left ventricular end-diastolic diameter (LVEDD, n ≥ 8 for each group), *P < 0.05 vs. NaCl; NS, not significant vs. NaCl. (D) Myocyte cross-sectional area as determined from 4 µm haematoxylin/eosin-stained sections in wild type and I-1-KO treated with NaCl or isoprenaline for 7 days (n > 260 cardiac myocytes from three to four hearts from each group, *P < 0.05 vs. NaCl). (E) Sirius red-stained paraffin sections representative for three to four hearts each. (F) Quantitative reverse transcriptase–polymerase chain reaction analysis of ANP mRNA concentrations in wild-type and I-1-KO hearts (n = 4 each), *P < 0.05 vs. NaCl. (G) Fractional area shortening (FAS) before and 10 min after injection of dobutamine (20 µg/g i.p; n = 15–21), *P < 0.05 vs. before dobutamine. (H) Quantitative reverse transcriptase–polymerase chain reaction analysis of I-1 mRNA concentrations in wild-type hearts treated with NaCl or isoprenaline (n = 4 each), *P < 0.05 vs. NaCl.
To test how I-1-KO respond to acute catecholamine administration after chronic isoprenaline treatment, we measured the effect of dobutamine on FAS in isoflurane anaesthesia. As shown before (Figure 3E), saline-treated I-1-KO and WT littermates showed a similar dobutamine-induced increase in FAS. Whereas this acute effect of dobutamine was completely absent in long-term isoprenaline-treated WT mice, it was partially preserved in identically treated I-1-KO (18 ± 5%, P < 0.05; Figure 5G). Finally, we determined I-1 mRNA levels by quantitative PCR. I-1 mRNA levels decreased by 50% after chronic isoprenaline infusion in WT (Figure 5H), supporting the idea that the decrease in I-1 in heart failure is secondary to chronic catecholamine stimulation.2,16,18
3.5 Inhibitor-1-deficient mice exhibit hypophosphorylation of ryanodine receptors and phospholamban
Given that the physiological functions of PP1 are not only controlled by its endogenous inhibitors I-1 and I-2, but to a large extent by its regulatory subunits,12 the consequences of I-1 ablation on the phosphorylation state of the various PP1c substrates are difficult to predict. We therefore performed western blots with antibodies directed against total and/or the phosphorylated forms of several known PP1c targets to identify alterations that may underlie the protective effect of I-1 ablation. We concentrated on (i) proteins involved in Ca2+ homeostasis (RyR2 and PLB), (ii) myofibrillar proteins (troponin-I and myosin-binding protein C), and (iii) nodal points of hypertrophic pathways likely affected by the β-adrenergic signalling pathway [glycogen synthase kinase 3β, protein kinase B (Akt), mitogen-activated kinases, and S6 ribosomal protein kinase]. The abundance and the phosphorylation level of most of these proteins were identical in I-1-KO and WT mouse hearts (Supplementary material online, Tables S3 and S4).
Interestingly, the changes in I-1-KO were confined to RyR2 and PLB, two major regulators of intracellular Ca2+ handling. The abundance and Ser2809 phosphorylation state (PKA site) of RyR2 was similar in I-1-KO and in WT (Figure 6A). In contrast, RyR2 Ser2815 phosphorylation (CaMKII site) was ∼30% lower in I-1-KO compared with WT (Figure 6A). Similarly, PLB phosphorylation at Ser16 (PKA site) was 30% lower in I-1-KO than in WT (Figure 6B). PLB phosphorylation at Thr17 (CaMKII site) showed similar trends but was not significantly different (0.75 ± 0.1 vs. 1.0 ± 0.1 in WT, n = 8, P = 0.1).
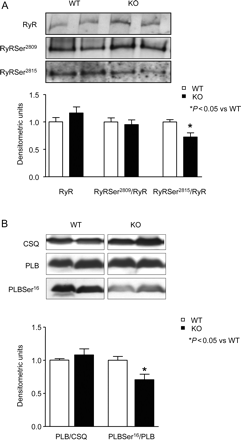
Hypophosphorylation of cardiac ryanodine receptor/Ca2+-release channel (RyR2) and phospholamban (PLB) in I-1-KO. (A) Western blot panels and quantification of total, PKA-, and CaMKII-phosphorylated RyR2 at Ser2809 and Ser2815, respectively, in wild-type (WT) and I-1-KO hearts (n ≥ 12 for each group). (B) Western blot panels and quantification of total, PKA-phosphorylated phospholamban at Ser16 in wild-type and I-1-KO hearts (n = 8 for each group).
4. Discussion
The present study demonstrates that the loss of I-1 in mice (i) leads to a mild reduction in cardiac β-adrenergic sensitivity without change in the maximal contractile response to acute β-adrenergic stimulation and without change in β-adrenoceptor density, (ii) protects from fatal catecholamine-induced arrhythmia without changing the heart rate response, and (iii) reduces the detrimental structural (hypertrophy, dilation, fibrosis), functional (loss of inotropic response to dobutamine), and molecular (increased ANP) effects of chronic β-adrenergic stimulation. These alterations were associated with lower steady-state phosphorylation levels of both RyR2 and PLB. The data indicate that the absence of I-1, and thereby of an intracellular amplification of β-adrenergic signals, is well compatible with life and an apparently normal phenotype, but, under conditions of increased adrenergic drive, is cardioprotective.
Conversely, I-1-TG mice with heart-specific overexpression of full-length I-1 developed spontaneous cardiac hypertrophy and mild cardiac dysfunction. I-1 appeared to accumulate in subsarcolemmal localization in these mice, contrasting our earlier observation of homogenous cytosolic distribution after adenovirus-mediated transfer of I-1 into cardiomyocyte monolayer cultures.15,26 Moreover, two independent lines of I-1-TG demonstrated a three-fold increase in PP1c protein abundance and activity which dominated under basal conditions as indicated by decreased PLB phosphorylation in isolated hearts. Thus, although β-adrenergic stimulation and subsequent PKA activation of overexpressed I-1 overcome PP1c (higher PLB phosphorylation in isolated hearts from I-1-TG), this clearly obscures cause–effect relations. Specifically, it is difficult to dissect whether the spontaneous hypertrophy observed in I-1-TG was due to the increase in I-1 itself and the expected decrease in PP1c activity under β-adrenergic stimulation or by the compensatory increase in PP1c abundance and activity. The latter explanation would be favoured by a similarly maladaptive phenotype of PP1c–TG mice.17 Other studies with TG mice overexpressing PP1c regulatory/targeting subunits such as inhibitor-223,24 or RGL/GM25 were also associated with an approximately seven-fold increase of PP1c protein levels, suggesting a stereotypic biological response.
I-1-KO mice had been investigated previously and were reported to have reduced basal contractility.17 We substantiated a mild subsensitivity of isolated cardiac preparations to β-adrenergic stimulation, well compatible with the amplifier role of I-1 in normal physiology. However, contractile function of the heart was shown by several means to be normal, both at the basal unstimulated state and after maximal β-adrenergic stimulation. Reasons for the discrepancy to the previous data are unknown, but could be related to differences in the depth of anaesthesia during echo and/or the degree of catecholaminergic stimulation. A mean heart rate of ∼600 b.p.m. in our experiments (Supplementary material online, Table S1) likely reflects minimal depressant effects of anaesthesia. Moreover, the absence of cardiac remodelling in I-1-KO for up to 1 (this study) or 1.5 years of age (former study17) also argues against a relevant abnormality of basal contractility in I-1-KO.
The most important new finding of the present study is that I-1-KO were significantly protected from the lethal consequences of isoprenaline in the presence of ketamine/xylazine anaesthesia and the adverse effects of a 7 day infusion of isoprenaline. What may account for the protection of I-1-KO compared with WT mice? A straightforward explanation is that cardiac myocytes from I-1-deficient mice show reduced sensitivity to β-adrenergic stimulation because they lack the I-1-mediated intracellular amplification loop. The consequence is that all cardiac responses to a given dose of β-adrenergic agonists (below maximal) will be smaller in I-1-KO than in WT. In this respect, I-1 deficiency acts as an ‘internal β-blocker’. However, Figure 4C shows that this is not exactly the case, because the heart rate-increasing effect of isoprenaline was superimposable in I-1-KO and WT, but markedly shifted to the right in the presence of the β-blocker metoprolol. Most likely, this difference is due to the fact that the β-blocker inhibits the entire signalling pathway, whereas I-1 deficiency affects signalling downstream of cAMP. cAMP mediates at least part of the heart rate-increasing effects of β-adrenergic stimulation directly by shifting the activation threshold of the hyperpolarization-activated cyclic nuclic nucleotide-gated ion channels to a less negative membrane potential.28
Another, likely related, explanation for the protective effect of I-1 ablation comes from the observation that RyR2 and PLB were hypophosphorylated in I-1-KO mice. This is important at least for two reasons. On the one hand, it substantiates in a whole-animal context that RyR2 and PLB are strongly controlled by PP1c and I-1, apparently more than the other PKA targets that were found to be unaffected (TnI, cMyBP-C, GSK3β, Akt, MAP kinases, and S6 ribosomal protein). On the other hand, alterations in RyR2 and PLB phosphorylation could be directly related to protection. RyR2 is part of a large macromolecular complex which is associated with proteins that regulate the channel function, including PKA, PP1 and PP2A, and the FK-506-binding protein FKBP12.6.7 Marks and colleagues7,8 showed that phosphorylation by either PKA (2809) or CaMKII (2815) increases RyR activity. In heart failure, RyR2 has been found to be hyperphosphorylated both at Ser2809 and Ser2815, which is associated with an increased channel-open probability and a Ca2+ ‘leak’ during diastole.7–9 Diastolic Ca2+ ‘leak’ via RyR2 channels is considered to contribute to impaired contractility, ventricular arrhythmias, and sudden cardiac death. Despite the ongoing debate on the true relevance of phosphorylation of the RyR2 in general and the PKA site in particular,29 the lower Ser2815 phosphorylation in I-1-KO is a strong candidate contributing to the protection from lethal arrhythmias and to the partial preservation of contractile response to dobutamine.
The second protein differentially phosphorylated in I-1-KO was PLB, which was hypophosphorylated at the PKA site. Since phosphorylation of PLB relieves its inhibitory effect on SERCA2a, the reduced phosphorylation state in I-1-KO should cause less efficient diastolic Ca2+ transport into the SR. This is generally considered an adverse effect because it could contribute to reduced diastolic relaxation, lower SR Ca2+ load, and therefore also lower systolic Ca2+ release. However, in combination with hypophosphorylation of RyR2 (and thereby reduced Ca2+ leak), hypophosphorylation of PLB may actually contribute to a new balance of SR Ca2+ release and Ca2+-reuptake at a lower level. This situation would mimic a lower degree of adrenergic stimulation and could be an attractive protective mechanism against arrhythmias and contractile dysfunction. At first sight, the concomitant affection of the PKA site of PLB and the CaMKII site of RyR2 in I-1-KO came as a surprise. Yet, it has been shown in a neuronal context that I-1 can mediate cross-talk between the PKA and CaMKII pathways by inhibiting the PP1-induced dephosphorylation (=inactivation) of CaMKII.30 Whether a similar pathway operates in cardiac myocytes is currently investigated.
The conclusions of the present study appear to be in contrast to those from a recent study showing that overexpression of truncated I-1 protected mice from contractile dysfunction in a pressure overload model.19 However, considerable differences exist in structure and function between WT and truncated I-1 (I-1ct): (i) I-1ct is constitutively active, i.e. does not need PKA-mediated phosphorylation for its activity towards PP131 and (ii) it lacks the PKC-phosphorylation sites of WT I-1 (Ser67 and Ser75) that decrease its activity towards PP1.14 Notably, the PKC-mediated regulation of I-1 has been associated with depression of cardiac contractile function13 and it is apparent that the Ser67 site was heavily phosphorylated in I-1-TG (Figure 1B). (iii) The affinity of I-1ct for PP1c is considerably lower than that of phosphorylated WT I-1.31 This feature could make I-1ct a ‘partial agonist’, stimulating the system (by inhibiting PP1c) at the basal state, but inhibiting it in situations of high catecholaminergic drive (by competing with phosphorylated endogenous I-1). (iv) Finally, evidence exists that I-1ct specifically acts on PLB without affecting RyR2 phosphorylation,19 which may be due to the lack of C-terminal sequences important for its subcellular distribution.32 These data in conjunction with the present study suggest that overexpressing I-1ct is not just functionally the opposite of ablating full-length I-1.
Our study has some limitations. First, the acute arrhythmia protocol is not well established and may not be directly transferable to chronic disease conditions. Ketamine, xylazine, and especially the combination are known to sensitize the heart to arrhythmogenic effects of catecholamines.33 We found a robust rightward shift of the dose–response curve for isoprenaline by applying the β-blocker metoprolol, indicating that the arrhythmias were indeed β-adrenoceptor-induced. Moreover, the reduction of RyR2 phosphorylation in I-1-KO is well compatible with current concepts of arrhythmogenic effects of β-adrenergic stimulation. Thus, we believe that the results are meaningful. A second limitation is that we searched for candidate proteins associated with the protection in I-1-KO and did not pursue an unbiased phospho-proteomic approach. However, given the moderate degree of difference between I-1-KO and WT, we estimate that it may be difficult to detect abnormalities in the protein or phosphorylation level of other proteins by using a less quantitative approach. Third, the interpretation of a complete, unconditional KO may be obscured by unknown compensations, but this limitation applies to all similar approaches. Finally, the protected phenotype of I-1-KO has only been tested under conditions of acute and 7 day catecholamine stress. It cannot be excluded that the lack of I-1 has detrimental consequences in situations in which a full acute fight and flight response is required.
The results presented here may have implications for the treatment of heart failure. Sympathetic over-activation is detrimental and accelerates the progression of heart failure.2 Conversely, β-adrenergic desensitization mechanisms and β-blockers are effective in patients with heart failure.34,35 I-1 ablation also appears to protect against adrenergic overstimulation without having apparent maladaptive consequences. This profile suggests therapeutic downregulation or inhibition of I-1 as a potential novel strategy for the treatment of heart failure.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study was supported by the Deutsche Forschungsgemeinschaft (DFG EL 270/3-1 to A.El-A. and T.E.) and by the European Union (EUGene Heart to T.E. and G.H.) and German Atrial Fibrillation Competence Network (grant 01Gi0204 to D.D.).
Acknowledgements
We thank Jutta Starbatty, Thomas Schulze, Birgit Geertz, Manja Schöne, and Annett Opitz for excellent technical assistance. I-1-KO mice were a kind gift from Drs Paul Allen and Paul Greengard (Rockefeller University, NY, USA).
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}