





Abstract
Cardiac sarcolemmal Na+–Ca2+ exchange is a central component of Ca2+ signaling essential for Ca2+ extrusion and contributing to a variable degree to the development of the systolic Ca2+ transient. Reports on differential gene expression of Na+–Ca2+ exchange in cardiac disease and the regulation of its thermodynamic equilibrium depending on intracellular gradients of ion concentrations between subcellular compartments have recently put a new complexion on Na+–Ca2+ exchange and its implications for excitation–contraction (E–C) coupling. Heart failure models and genetic approaches to regulate expression of the Na+–Ca2+ exchanger have improved our knowledge of exchanger function. Modest overexpression of the Na+–Ca2+ exchanger in heterozygous transgenic mice had minimal effects on E–C coupling and cardiac function. However, higher levels of Na+–Ca2+ exchange expression in homozygotes led to pathological hypertrophy and failure with an increased interaction between the L-type Ca2+ current and Na+–Ca2+ exchange and reduced E–C coupling gain. These results suggested that the Na+–Ca2+ exchanger is capable of modulating sarcoplasmic Ca2+ handling and at high expression levels may interact with the gating kinetics of the L-type Ca2+ current by means of regulating subsarcolemmal Ca2+ levels. Despite being a central component in the regulation of cardiac E–C coupling, a newly generated mouse model with cardiac-specific conditional knock-out of the Na+–Ca2+ exchanger is viable with unchanged Ca2+ dynamics in adult ventricular myocytes. Cardiac myocytes adapt well to knock-out of the exchanger, apparently by reducing transsarcolemmal fluxes of Ca2+ and increasing E–C coupling gain possibly mediated by changes in submembrane Ca2+ levels. For E–C coupling in the murine model, which relies primarily on sarcoplasmic Ca2+ regulation, this led to the suggestion that the role of Na+–Ca2+ exchange should be thought of as a Ca2+ buffering function and not as a major Ca2+ transporter in competition with the sarcoplasmic reticulum.
1. Regulation of Na+–Ca2+ exchange in microdomains for Na+ and Ca2+
Upon depolarization of the plasma membrane, Ca2+ influx through the opening of voltage-gated L-type Ca2+ channels triggers Ca2+ release from the terminal cisternae of the junctional sarcoplasmic reticulum (SR) [1]. The high levels of Ca2+ released by the SR rapidly diffuse down a steep intracellular concentration gradient to the contractile filaments and induce contraction of the myocyte. Thus Ca2+-induced Ca2+ release can initiate and fine-tune the conversion of the electrical signal of membrane excitation into the mechanical response of myocardial contraction and is critical for the regulation of excitation–contraction (E–C) coupling. Entry of Ca2+ with each contraction requires an equal amount of Ca2+ extrusion within a single heartbeat to maintain Ca2+ homeostasis and to ensure relaxation. The principal cardiac extrusion mechanism is Na+–Ca2+ exchange (NCX). Recent studies suggest that NCX is also directly involved in the regulation of E–C coupling by means of modulating SR Ca2+ load [2], SR Ca2+ release [3–5] and Ca2+ spark frequency [6].
In the present manuscript we will review the experimental evidence for a possible involvement of NCX in Ca2+ regulation and E–C coupling. We will focus on the quantitative and spatial arrangements of Ca2+ handling proteins and their dependence on putative submembrane gradients of Ca2+ and Na+.
1.1. Physiology of Na+–Ca2+ exchange
The cardiac sarcolemmal NCX utilizes the Na+ electrochemical gradient to mediate the electrogenic countertransport of 3 Na+ ions for 1 Ca2+ ion across the sarcolemmal membrane [7] This stoichiometry is widely accepted although some later reports suggested a ratio of 2:1 [8] or 4:1 [9,10]. Exchange is bidirectional and capable of moving Ca2+ in either direction across the sarcolemma. The exchanger will thereby always tend to bring cytoplasmic free Ca2+ to its thermodynamic equilibrium, which depends primarily on membrane potential and the Na+ gradient. With a KD of 5–6 μM, the affinity of the exchanger for [Ca2+]i is more than an order of magnitude higher than the affinity for [Ca2+]o. The apparent affinity for intracellular Na+ is around 10–20 mM, about four to five times less than for [Na+]o. Besides being the substrates for exchange activity, Na+ and Ca2+ have also been shown to exert a regulatory function on the exchanger. At a high affinity binding site located on the large intracellular loop, regulatory Ca2+ can strongly modulate exchange activity by rapid allosteric activation at an apparent affinity of 50–125 nM. Exposure of the intracellular surface to high levels of Na+ results in time-dependent inactivation of NCX to a new steady-state level of exchange activity [11]. This inactivated state can be modeled to arise from the fully Na+-loaded exchanger molecule, when three Na+ ions bind to the transport sites at the intracellular surface of the protein and therefore has a third-power dependence on cytoplasmic Na+. Other physiological regulators of NCX include the signaling lipid phosphatidylinositol-4,5-bisphosphate (PIP2), free radicals, pH, and temperature as well as protein kinases with stimulation of exchange activity by agents acting via protein kinase C (reviewed in [12]). However, proof is still lacking for the physiological significance of these messengers as regulators of NCX in vivo, especially in cardiac cells.
1.2. Regulation of ion activity and exchanger function in the ‘fuzzy space’
The concept of subcellular compartmentation [13,14] has given a new complexion to exchanger function and its dependence on ion activity and transmembrane gradients of [Ca2+] and [Na+]. The existence of a microdomain or ‘fuzzy’ space for Ca2+ between the junctional SR and the cytoplasmic leaflet of the T-tubular sarcolemmal membrane is generally accepted and has received strong experimental support by the discovery of calcium sparks with limited spatial spread [15] and by the relative contributions of Ca2+ influx via Ca2+ channels and NCX to the activation of ryanodine receptors [3,5,16]. At a size of 50–200 nm across and a depth of 15 nm for each compartment [17,18] resulting in a volume of ∼8 × 10−20 l each and approximately 10000 of these microdomains per cell, depending on species and cell size the total volume of this so-called diadic cleft space would correspond to ∼0.1% of total cell volume. In this restricted space the presence of a large number of sarcolemmal binding sites for Ca2+[18] will further limit diffusion and reduce the stochastic probability of free Ca2+ ions within the diadic cleft [18]. In a detailed model of the time course of [Ca2+] in this microdomain Langer and Peskoff [19] calculated that during a 0.3 pA L-type Ca2+ current of 1 ms duration peak [Ca2+] would rise to >1 mM. Furthermore the strong sarcolemmal buffering resulted in a slow decay of Ca2+ levels with [Ca2+] still over 10 μM after 10 ms [19]. However, ion activity may further be limited due to the electric field of excitable membranes which cause Ca2+ to accumulate near the sarcolemma, building substantial gradients in [Ca2+] between SR and sarcolemma. Including these electrostatic effects on ion movements Soeller and Cannell [17] predicted more rapid changes in Ca2+ in response to Ca2+-induced Ca2+ release but lower peak [Ca2+] levels of ∼300 μM at the center of the diad gradually decreasing to ∼45 μM at the edge. These theoretical considerations are of relevance regarding the dependence of the driving force of NCX on ion concentrations since it is ion activity rather than concentration that thermodynamically matters [20].
Efficient Ca2+-induced Ca2+ release is restricted to the diadic cleft space in the T-tubular regions. Ultrastructural data indicate that NCX is also most abundant in the T-tubular membranes [21]. This was recently confirmed by functional studies using de-tubulation, which indicated that most of the NCX current originated from the T-tubular compartment and that the exchanger could be co-localized here with the Na+/K+ pump at a similar high densities [22,23]. Co-localization studies within the T-tubules using high resolution imaging, however, modeled NCX outside of the diadic cleft, showing that the degree of co-localization of ryanodine receptors with the L-type Ca2+ channels was significantly higher than with NCX [24]. A possible involvement of NCX in the regulation of local [Ca2+] within the diadic cleft was proposed by the observation that activation of the Na+ channels in the presence of Ca2+ channel block triggered Ca2+ release from the SR [3,13]. However, systolic Na+ influx via INa is only expected to raise global [Na+]i by 10 to 15 μmol/l (≈0.1%; [25]) and would thereby not be sufficient to induce reverse mode NCX and Ca2+ influx by the order of magnitude required to activate the ryanodine receptors.
Following the idea of subcellular compartmentation this observation led to the proposal of a subsarcolemmal ‘fuzzy’ space for [Na+] which may not strictly coincide with the Ca2+ microdomain [14] (Fig. 1). Experimental evidence for a restricted diffusion space for Na+ has been derived from the analysis of the current of Na+-dependent transporters like the Na+-pump [26], the Na+-activated K+ current [27] or NCX [28]. Based on these measurements the subsarcolemmal compartment for Na+ was calculated to comprise between 1 and 14% of total cytosolic volume [26,28,29]). Differences of these measurements may be due to individual experimental approaches but may also reflect a marked microheterogeneity for [Na+]i that was observed by X-ray microprobe analysis of subsarcolemmal Na+ gradients in guinea-pig ventricular myocytes [27]. Despite these quantitative differences, all of these measurements strongly suggest that the size of the calculated restricted space for Na+ exceeds diadic cleft volume.
![Theoretical modeling of subsarcolemmal ion gradients in the diadic cleft and the ‘fuzzy’ space for [Na+] under physiological conditions (A) and in the presence of higher sarcolemmal densities of NCX (B). The Ca2+ micordomain or diadic cleft space spans between the T-tubular membrane with high densities of L-type Ca2+ channels (ICa) and the terminal cisternae of the sarcoplasmic reticulum with the ryanodine receptors (RyR). Efficient Ca2+-induced Ca2+ release is restricted to the diadic cleft space indicated by the darkly shaded area. The microdomain for [Na+] (lighter shaded area) extends beyond the diadic cleft and contains the Na+–Ca2+exchanger (NCX), the Na+/K+–ATPase (ATP) and presumably the Na+-channels (INa). Higher expression levels of NCX (B)may result in closer proximity of the exchanger to ICa with altered Ca2+ handling and defects in E–C coupling as suggested by the reduced gain in a transgenic mouse model with 3.1-fold overexpression of NCX [48].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/67/2/10.1016/j.cardiores.2005.04.031/2/m_67-2-198-fig1.gif?Expires=1716530999&Signature=AKDBdvA-FXEs2rk6gG8OIQs7-HIRxAsCGYQHpc7b1Xkr~FHX4y7gDf2IWv81swJPPIFMrL1OSwvxNyqO5MkUQwmc2Gx~dJop7myuAEGRwfOvU0NfD8zZpc-sq77CSVqZgT2dm7iZT85wJgUUuaeYptqeq5~IVtQ8-jdoRWtCf45Y-tEhRqfeRYOoMkWs9X-K5K6ECUlX1URmG2z16b1e4wahN-Ugh9hXlslF97sJVKqLYgtc8CRlpRThDjIURU3M4UoZTOYbSoJga8Dhe9PZFq0UnxZo1zHLH-2FHVndZTcPqjx8E3ycPb38a-b82bD9fbt2uZI6cGGpEJlRokx1hQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Theoretical modeling of subsarcolemmal ion gradients in the diadic cleft and the ‘fuzzy’ space for [Na+] under physiological conditions (A) and in the presence of higher sarcolemmal densities of NCX (B). The Ca2+ micordomain or diadic cleft space spans between the T-tubular membrane with high densities of L-type Ca2+ channels (ICa) and the terminal cisternae of the sarcoplasmic reticulum with the ryanodine receptors (RyR). Efficient Ca2+-induced Ca2+ release is restricted to the diadic cleft space indicated by the darkly shaded area. The microdomain for [Na+] (lighter shaded area) extends beyond the diadic cleft and contains the Na+–Ca2+exchanger (NCX), the Na+/K+–ATPase (ATP) and presumably the Na+-channels (INa). Higher expression levels of NCX (B)may result in closer proximity of the exchanger to ICa with altered Ca2+ handling and defects in E–C coupling as suggested by the reduced gain in a transgenic mouse model with 3.1-fold overexpression of NCX [48].
Although the first observations of significant Ca2+ release by reverse NCX remain controversial as they could only be reproduced by some [4,5], but not by others [16,30], the concept of a Na+ microdomain has stimulated extensive research and theoretical modeling. Using different values of [Na+]i, Bers suggested that just a 3 mM difference in cytosolic Na+ could lead to a significant difference in NCX-mediated Ca2+ influx. In rabbit cardiac myocytes, he predicted that with [Na+]i of 10 mM, Ca2+ influx by NCX occurred during most of the action potential but that at [Na+]i of 7 mM, this occurred only in the initial phase following depolarisation [31]. And more recently a model study by Han et al. demonstrated a steep rise in SR triggering efficiency to about 25% by sodium–calcium exchange in response to an increase in [Na+]i to 10 mM [32]. The impact of cytoplasmic Na+ on the thermodynamic equilibrium is quantitatively important because it contributes to the driving force of the exchanger with approximately the third power. Thus the variability of intracellular Na+ concentrations among species [33] as well as elevated Na+ levels under pathophysiological conditions like heart failure [34,35] or hypoxia [36] may be critical for the efficacy of NCX to trigger Ca2+ release from the SR.
But the reactions of the exchanger to changes in [Na+]i are more complex since binding of Na+ to cytoplasmic transport sites in the reverse mode also leads to inactivation of the exchanger. This inactivation process, however, is strongly dependent on cytoplasmic pH and temperature, is eliminated in the presence of increasing levels of cytoplasmic regulatory Ca2+ and takes several seconds under experimental conditions to reach a new steady-state level of exchange activity (τ = 4.4 s; [11]).
Regardless of its impact on Ca2+ release from the SR, it is well accepted that the decreased electrochemical potential of Na+ across the sarcolemma caused by the increased [Na+]i reduces the forward driving force of NCX, i.e., Ca2+ extrusion.
In different models of hypertrophy and heart failure, increased densities of NCX in the plasma membrane have been reported [37–39] and even chronic sympathetic activation by itself may act as a stimulus in the regulation of NCX expression [40]. Changes in the density and the spatial distribution of NCX in the plasma membrane are relevant to the timing and the momentary magnitudes of the exchange current in response to modulations of its thermodynamic equilibrium. Different animal models with increased and reduced levels of expression of NCX have been employed to study these effects on E–C coupling and we will review their results in the following sections.
2. Ca2+ handling and contractility in the presence of increased Na+–Ca2+ exchanger densities
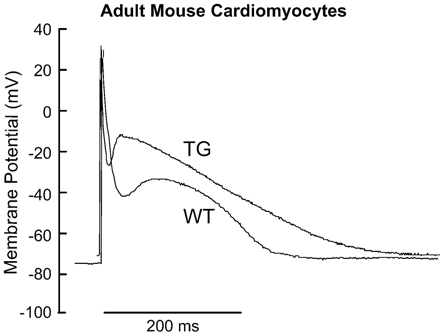
The transgenic mouse with cardiac-specific overexpression of the canine NCX has widely been employed to study myocardial Ca2+ regulation by this countertransporter both under physiological and pathological conditions (reviewed in [41]). Initial studies of voltage-clamped isolated myocytes from these animals demonstrated that forward function of the exchanger was increased 2.3-fold in heterozygous mice and 3.1-fold in homozygous overexpressors. Despite this enhancement in transsarcolemmal NCX, adaptations in the expression level of other Ca2+ handling proteins have not been detected. Thus it appears that the faster Ca2+ transients and twitches displayed by the transgenic myocytes in comparison to their wild type littermates [42] are mainly attributable to the high density of NCX. Typical action potentials recorded from isolated cardiac myocytes of homozygous NCX overexpressing mice and wild type controls are shown in Fig. 2. Action potentials from transgenic myocytes had a lower peak amplitude, a higher plateau and a delayed terminal repolarization. These characteristics are consistent with the idea that the outward current via NCX in overexpressors is stronger during the upstroke of the action potential causing the decrease in peak amplitude, while the higher plateau and prolonged APD90 represent in part an enhanced forward function of NCX during the course of the Ca2+ transient. Compared to wild type these transgenic animals showed no change in [Na+]i that would influence exchange activity [11].
| RMP (mV) | APA (mV) | Plateau (mV) | APD50 (ms) | APD90 (ms) | |
|---|---|---|---|---|---|
| WT n = 6 | 73.1 ± 3.1 | 86.4 ± 7.6 | 40.1 ± 1.8 | 15.6 ± 2.0 | 210.0 ± 13.4 |
| TG n = 8 | 71.6 ± 2.4 | 74.4 ± 5.9 | 53.7 ± 2.5 | 11.3 ± 1.3 | 316.7 ± 15.4 |
| p (WT vs. TG) | 0.607 | 0.040 | 0.018 | 0.082 | <0.0001 |
| RMP (mV) | APA (mV) | Plateau (mV) | APD50 (ms) | APD90 (ms) | |
|---|---|---|---|---|---|
| WT n = 6 | 73.1 ± 3.1 | 86.4 ± 7.6 | 40.1 ± 1.8 | 15.6 ± 2.0 | 210.0 ± 13.4 |
| TG n = 8 | 71.6 ± 2.4 | 74.4 ± 5.9 | 53.7 ± 2.5 | 11.3 ± 1.3 | 316.7 ± 15.4 |
| p (WT vs. TG) | 0.607 | 0.040 | 0.018 | 0.082 | <0.0001 |
Original recordings of action potentials of isolated ventricular myocytes from homozygous Na+–Ca2+ exchanger overexpressing mice (TG) and wild type controls (WT). Resting membrane potentials (RMP) were similar in myocytes of the two groups. However, compared to the WT, myocytes from TG show a significant reduction in peak action potential amplitude (APA) consistent with faster initial repolarisation likely mediated by enhanced reverse mode Na+–Ca2+ exchange. Action potentials from transgenic myocytes also presented a higher plateau and delayed terminal repolarisation (APD90) indicating that whole cell inward current is increased during the course of the Ca2+ transient most likely supported by an enhanced forward mode Na+–Ca2+ exchange and increased L-type Ca2+ current.
2.1. Regulation of SR Ca2+ handling by Na+–Ca2+ exchange
In these hypercontractile myocytes, pharmacological inhibition of SERCA activity with thapsigargin was employed to test whether overexpression of NCX can compensate for a reduction in SERCA function that may occur under pathophysiological conditions [42]. It could be shown that a 2.3-fold increase in NCX activity in ventricular myocytes from heterozygous animals compensated for a reduction of SERCA function by 28% so maintaining the characteristics of the Ca2+ transient from wild type hearts. These data confirm that, to some extent, NCX can compensate for compromised SERCA function and allow Ca2+ homeostasis to be maintained in the mouse heart.
Compelling evidence for the efficiency of reverse NCX to trigger Ca2+ release from internal stores in this model was provided by Yao et al. examining the effects of nifedipine on intracellular Ca2+ transients in field stimulated isolated myocytes [33]. In the presence of 10 μM nifedipine Ca2+ transients were preserved for over 5 minutes in ventricular myocytes from NCX overexpressors but not in their wild type littermates. However, the time to peak was markedly prolonged in the nifedipine-treated transients indicating that NCX operating in reverse mode is less efficient for triggering SR Ca2+ release as compared to physiological Ca2+ currents.
Enhanced Ca2+ entry into the cytosol via NCX has also been seen as the likely mechanism to produce a larger SR Ca2+ content in heterozygous overexpressors as compared to controls. Using a protocol for paired rapid cooling contractures in heterozygous NCX overexpressing cardiomyocytes, Terracciano et al. observed that during rewarming and at rest more Ca2+ was taken back up into the SR than that expelled from the SR during the first cooling. Since this extra SR loading was inhibited by Na+-and Ca2+-free superfusate, they concluded that Ca2+ entry via NCX may occur during the decline of intracellular Ca2+ in the late phase of action potential or even at rest [43].
In other species acute overexpression of NCX was induced by adenoviral gene transfer [44–46]. In rabbit myocardium increased NCX resulted in depression of contractility as well as systolic and diastolic Ca2+ levels at any stimulation frequency as shown by a blunted force-frequency relationship [44,46]. These observations support the notion that upregulation of NCX primarily depletes SR Ca2+ stores resulting in systolic myocardial failure. In rat myocardium, however, we found a significant increase of peak Ca2+ and fractional shortening at low stimulation rates in isolated myocytes upon adenoviral gene transfer emphasizing the role of the beating rate and of [Na+]i in regulating the predominant direction of Ca2+ transport via NCX in the course of the physiological action potential [45].
2.2. Kinetics of ICa in the presence of Na+–Ca2+ exchange overexpression
The L-type Ca2+ current (ICa) is the main physiological trigger for SR Ca2+ release. Cardiac ICa is rapidly activated by membrane depolarization while subsequent inactivation follows kinetics dependent on time, membrane potential and [Ca2+]i. The Ca2+-dependent inactivation of ICa is likely mediated by calmodulin bound to the α1C subunit of the channel and as such may provide a feedback control to limit excessive Ca2+ entry [47].
The term gain of E–C coupling is referred to as the amplification factor of Ca2+-induced Ca2+ release and can be expressed as the ratio of [Ca2+] released by the SR and the peak Ca2+ current to account for the observation that in most cases only the first few milliseconds of the calcium current are involved in triggering Ca2+ release. Despite unchanged SR Ca2+ load, we noted a significant reduction in the Ca2+ transient amplitude in isolated mouse myocytes homozygous for NCX overexpression [48]. On the other hand, peak ICa in these cells was larger which consequently resulted in a significant reduction in the gain of E–C coupling for these homozygous animals. The increase of the L-type Ca2+ current was especially apparent at negative depolarizing potentials (−20 to 0 mV, 0 mM Na+ in pipette solution) where NCX would strongly be operating in the forward mode (Fig. 3), extruding Ca2+ from the cytosolic environment. Strong evidence for a possible impact of INa–Ca in the regulation of L-type Ca2+ influx was provided by transiently blocking the exchanger in Na+-free bath solution. With no exchange activity, the L-type current could be significantly reduced. At more positive membrane potentials, blocking the exchanger by removal of bath Na+ did not affect the peak Ca2+ current but still resulted in an increase of the intracellular Ca2+ transient. This increase in Ca2+ release flux associated with blockade of NCX at +10 mV is consistent with the idea that the overexpressed exchanger population may act as a sink for Ca2+, reducing trigger Ca2+ flux before the ryanodine receptors sense that Ca2+. As previously shown by others [3–5], NCX appears to be capable of modulating SR Ca2+ release, presumably by altering the concentration of Ca2+ in subsarcolemmal regions. Ca2+-induced inactivation of ICa is influenced predominantly by Ca2+ released by the SR [49], however the removal of Ca2+ from the diadic cleft by NCX may have an additional impact on the kinetics of the L-type current represented by the increased peak current and the slowed inactivation of ICa with a prolonged open time τ[48].
![L-type Ca2+ current in isolated ventricular mouse myocytes from wildtype (▪), NCX1 knockout (▵) and transgenic homozygous mice overexpressing NCX1 (▾). Single cells were patch-clamped and depolarized from a holding potential of −40 mV to a family of test potentials from −30 to +40 mV. Note that ICa is decreased at all potentials in the knockout whereas it is increased in the overexpressors as compared to the wildtype. * p<0.05 vs. wildtype at indicated voltage; †p<0.05 vs. wildtype by 2-way ANOVA. Modified from Reuter et al.[48] (wildtype and overexpressor) and Henderson et al.[73] (knockout).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/67/2/10.1016/j.cardiores.2005.04.031/2/m_67-2-198-fig3.gif?Expires=1716530999&Signature=AQ0FfoERCs2U4x5RNfnVX250OpWKQk08oU4LJzjv772LcxSjnw1k1~wyX8N-MnqfGSJBE-lWnhmvgua-mJ5byyemEcuOgoAEI906kXyiErNlrj1E3RywDGcgQlA71G1LMFNk-CGMrmjFN8rtoAKvv3hU17LLIB5kr31QxVmqLHK2FARsPHnkDsu1inCgh0UmJZi-xSkNrAtK41hyl17jqWKsGuONcM7X87RLDXA3i8KU-17v7BDQN62xXaMxRMmnuXiqRbsc5iqZktefqdw39GPVy7O2idHO0spyVSTXYD-mrfdD1cvqfDdcVpA4APZdOI9g6DNpSF7MerCoT7pdfw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
L-type Ca2+ current in isolated ventricular mouse myocytes from wildtype (▪), NCX1 knockout (▵) and transgenic homozygous mice overexpressing NCX1 (▾). Single cells were patch-clamped and depolarized from a holding potential of −40 mV to a family of test potentials from −30 to +40 mV. Note that ICa is decreased at all potentials in the knockout whereas it is increased in the overexpressors as compared to the wildtype. * p<0.05 vs. wildtype at indicated voltage; †p<0.05 vs. wildtype by 2-way ANOVA. Modified from Reuter et al.[48] (wildtype and overexpressor) and Henderson et al.[73] (knockout).
The dependence of microscopic gain on the open time τ of the L-type Ca2+ channel has recently been modeled by Soeller and Cannell [17] based on gain curves redrawn from several studies of integrative local-control models. Using a simplified model of Ca2+ release by single L-type channel currents in the diadic cleft they showed a strong dependency of gain on τ with a maximum occurring near physiological values of τ. Soeller and Cannell concluded that if gain could be increased by altering L-type channel open times it might be possible to restore contractility in animal models of heart failure with depressed microscopic gain. Thus, the increased open time and peak Ca2+ current of the L-type Ca2+ channel may help support near normal Ca2+ release in this model.
Reductions in gain underlying defects in E–C-coupling have been demonstrated previously in heart failure [50] and even in human failing myocardium Piacentino III et al. [51] showed a tendency of the current–voltage (I/V) relationship of ICa to increased peak currents and a leftward shift of the I/V relationship analogous to our observation. Changes in the spatial arrangement of SR Ca2+-release channels and the L-type Ca2+ channels have been invoked as possible explanation for these alterations of E–C coupling [50] and other compensatory changes in the response of ryanodine receptors to trigger Ca could modify the function of gain as well. Based on our measurements in the homozygous transgenic mouse model we speculate that the increased density of NCX directly modifies Ca2+ levels in subsarcolemmal regions including the diadic cleft space (Fig. 1). Additional exchangers packed into the sarcolemmal membrane may result in a closer proximity to L-type Ca2+ channels. This could increase the direct impact of NCX on Ca2+–induced Ca2+ release and L–type Ca2+ channel gating or could at least aggravate defects in E–C coupling resulting from possible structural changes or altered channel gating.
2.3. Na+–Ca2+ exchange overexpression in heart failure
The failing heart shows by definition an impaired contractile function resulting in the inability to provide sufficient cardiac output adequate for the metabolic needs of the organism. The contractile deficit of the failing heart present at low beating rates becomes even more apparent when the heart is challenged by increasing frequencies of stimulation resulting typically in a less positive or even negative force–frequency relationship [52]. To date it is widely accepted that this impaired contractility is due to an altered Ca2+ homeostasis in failing cardiac cells [53]. Among the many functional and molecular changes identified in the failing heart [54], the impaired function of the SR as an intracellular Ca2+ store seems to be of critical importance. A reduced activity and expression of the SERCA [55,56] and an abnormal gating mechanism of the release channel [57] may result in a decreased SR Ca2+ content in failing cardiac myocytes [58] with the lack of a frequency-dependent upregulation present in nonfailing myocardium [59].
Hypertrophy and heart failure is often associated with an increased cell size. The respective decrease in the surface to volume ratio would seem to require the upreglation of sarcolemmal transporters like NCX in order to maintain Ca2+ homeostasis. However, in view of the compromised Ca2+ accumulation within the SR alterations in the expression level of NCX appear to play a critical role for E–C coupling in heart failure. On one hand, elevated densities of NCX protein [37,38] with an approximate 87% increase in exchange activity [39] observed in heart failure could exacerbate systolic dysfunction by cellular Ca2+ depletion. This view is supported by transient overexpression of NCX in rabbit ventricular myocytes described above, where the phenotype showed depressed contractility and reduced SR Ca2+ content [44,46]. On the other hand, Ca2+ influx via reverse mode NCX could support the development of the systolic Ca2+ transient in the setting of a compromised SR function. In an elegant study on failing human hearts correlating frequency potentiation of systolic and diastolic tension with protein expression Hasenfuss et al. [60] consistently found an impaired systolic function and the ratio in protein expression of NCX to SERCA to be increased by a factor of 2 to 4 compared to nonfailing myocardium. By discriminating the myocardial tissue according to their diastolic function they identified different phenotypes ranging from hearts with increased protein levels of NCX and unchanged SERCA levels to myocardium with decreased SERCA expression and unchanged levels of NCX. Although the ratio of the two proteins competing for cytosolic Ca2+ was comparable in all groups, diastolic function and frequency-dependent force generation positively correlated with the level of expression of the exchanger protein.
3. Excitation–contraction coupling under reduced Na+–Ca2+ exchange
During relaxation there is a dynamic competition for the cytosolic Ca2+ among SERCA>NCX>sarcolemmal Ca2+ pump ≥ mitochondria, according to their quantitative contribution. Depending on species and condition, NCX contributes a variable amount toward [Ca2+]i decline during twitches ranging from 9% in rat [61] and mouse [62] to 29% in ferret [63]. While studies on different species have demonstrated that the amount of systolic Ca2+ extruded by the sarcolemmal Ca2+ pump is consistently under 5% it has been hypothesized that it may be especially important in the regulation of diastolic Ca2+ efflux since it can have a higher Ca2+ affinity (Km(Ca2+)=64 nM) upon stimulation with calmodulin [64] than NCX. The lack of highly selective inhibitors of NCX [65] or the sarcolemmal Ca2+ pump have made it difficult to study cardiac E–C coupling under physiological conditions in the absence of either of these two Ca2+ lowering mechanisms. Different approaches to reduce function and expression of the cardiac NCX by genetic manipulation have recently put a new complexion on NCX and its implications for E–C coupling.
3.1. Antisense Inhibition of Na+–Ca2+ exchange expression
Antisense inhibition of NCX in adult rat myocytes resulted in knock-down of exchanger protein by 30% after 3 days without proven adaptations in the expression level of other Ca2+ handling proteins [66]. Under physiological conditions (i.e., 1.1 mM [Ca2+]o, 37 °C, stimulation at 1 and 3 Hz) this mild reduction in NCX showed a significant increase in resting Ca2+ levels but had no effect on the development of the intracellular Ca2+ transient, contractility, or morphology of the action potential. However, the responses in contractility and [Ca2+]i of these cardiomyocytes when exposed to low [Ca2+]o (0.6 mM) or increased extracellular Ca2+ at 5 mM were diminished compared to controls. These experiments demonstrated that mild changes of exchanger density simply alter the momentary magnitudes of the exchange current. Extracellular Ca2+ is a relatively strong determinant of the exchanger equilibrium and the magnitudes of Ca2+ efflux and influx following changes in [Ca2+]o were decreased, respectively in the presence of reduced levels of exchanger expression.
3.2. Inactivation of Na+–Ca2+ exchange by global knock-out
Global knock-out of NCX in the mouse was reported by four laboratories, including ours, to be embryonic lethal at about 11 days post coitum [67–70]. Despite structural defects [67,68], heart tubes at 9.5 days post coitum showed almost normal Ca2+ dynamics under low stress conditions (Fig. 4) with regional contractions both spontaneously and upon electrical stimulation [68]. Although protein expression and function of the sarcolemmal Ca2+ pump were similar in heart tubes from knock-out and control embryos it was proposed that this alternative mechanism for Ca2+ extrusion compensated for the loss of exchanger function under these conditions. However, this mechanism seemed not to be sufficient when any intervention was used that increased the need for Ca2+ efflux, e.g., increased stimulation frequency, application of caffeine or isoproterenol [68]. These morphological and functional characteristics suggested that lethality of the embryos was of cardiac origin. A recent attempt to rescue the mutant embryos by transgenic re-expression of cardiac NCX increased survival only by one day [71].
![Ca2+transients in heart tubes from embryos with global Na+–Ca2+ exchanger knock-out compared to wild type and their response to the replacement of 140 mM Na+o with Li+o. With external field stimulation at 1 Hz, heart tubes in both groups had similar kinetics for the rise and decay of Ca2+ transients under control conditions (left). In heart tubes from wild type embryos (upper panel), replacement of 140 mM Na+o with Li+o developed Ca2+ overload within 1 minute. Wash out of extracellular Na+ had no effect on Ca2+ transients in heart tubes from Na+–Ca2+ exchanger knock-outs over at least 5 minutes (lower panel). Stimulation voltage was increased from 30 to 60 V to retain excitability. Reproduced from Reuter et al.[75].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/cardiovascres/67/2/10.1016/j.cardiores.2005.04.031/2/m_67-2-198-fig4.gif?Expires=1716530999&Signature=ATXgmtieNLwezmH7deWK5rwLEVKhipfX0DRsFEJt3abY60-trJoS2jA1hgyT4zN770e7CDC6xhHXrD7YpgbX4oNNFSjkPTNSACQw9Umw2xG2hvpdadjAjNQByJ65JAeG8mpLvpEcKioxlEFUyAs7AbIhHmjmovyDGZjW8L~Ig69tH8F~2qU3ygQwgUNj5BhryHzZuaLOHgw87d3kWToPLXphNn7de6L5YkkvHENp~JE5jeR08lfsiQTOkfUAeEcVYgrTEC931Fep6iX1~ABSlHkZbNGr9wcbqrMHylz72qnEMw4~WRbyaZN-BBsCPZa77qrgC6tUmGOB0i1WzQgdSQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ca2+transients in heart tubes from embryos with global Na+–Ca2+ exchanger knock-out compared to wild type and their response to the replacement of 140 mM Na+o with Li+o. With external field stimulation at 1 Hz, heart tubes in both groups had similar kinetics for the rise and decay of Ca2+ transients under control conditions (left). In heart tubes from wild type embryos (upper panel), replacement of 140 mM Na+o with Li+o developed Ca2+ overload within 1 minute. Wash out of extracellular Na+ had no effect on Ca2+ transients in heart tubes from Na+–Ca2+ exchanger knock-outs over at least 5 minutes (lower panel). Stimulation voltage was increased from 30 to 60 V to retain excitability. Reproduced from Reuter et al.[75].
Embryonic cardiomyocytes do not represent the ideal system for studying the role of NCX on E–C coupling. Besides being a difficult preparation not suitable for voltage clamp analysis, internal Ca2+ stores are poorly developed at this early developmental stage and intracellular Ca2+ dynamics are primarily dependent on transsarcolemmal Ca2+ fluxes [72]. Nevertheless, these data presented new roles of the exchanger in embryogenesis and suggested that the sarcolemmal Ca2+ pump could possibly serve as a backup system for NCX.
3.3. Functional adult myocardium in the absence of Na+–Ca2+ exchange
A more detailed picture of the role of the exchanger in the regulation of the murine E–C cycle was recently presented by a study on mutant mice with a cardiac-specific knock-out of NCX [73]. Strikingly, these mice lived to adulthood with only a modestly reduced contractility of 20–30% as assessed by echocardiography at 7.5 weeks of age. What would have been inconceivable until then was the finding of unchanged Ca2+ dynamics in externally paced isolated adult ventricular myocytes despite the absence of NCX with systolic and diastolic Ca2+ levels similar to control cells and no relaxation deficit. During prolonged caffeine application there was very limited Ca2+ efflux indicating that no alternative efflux mechanism, as for example the sarcolemmal Ca2+ pump, compensated for the absence of the exchanger.
The murine model has short action potentials and shows a distinct pattern of Ca2+ flux pathways that seems to be favorable for a system to study E–C coupling in the absence of NCX. With only 9% of activator Ca2+ crossing the sarcolemma during each contraction cycle, the contribution of transsarcolemmal Ca2+ fluxes to initiate E–C coupling is small in mice and rats as compared to most other species [61,62]. Under these conditions NCX will have relatively little impact on the magnitude of the intracellular Ca2+ transient during the regular E–C cycle, however, it can be predicted that even small changes to its thermodynamic equilibrium would have strong effects on E–C coupling. This led Hilgemann to the suggestion that its role in this respect should be thought of as a Ca2+ buffering function and not as a major Ca2+ transporter in competition with the SERCA [74]. To test this theory Hilgemann simulated the effect that small changes of some parameters that set the exchanger equilibrium ([Na+]i, [Ca2+]o) as well as of major Ca2+ handling proteins (SERCA, Ca2+ channel, SR release or NCX) have on the peak Ca2+ level achieved on activation. Based on this simulation he showed that changes in [Na+]i and in SERCA function had the largest influence on peak Ca2+. Interestingly, changes of [Ca2+]o resulted in proportional changes of peak intracellular Ca2+ whereas changes of the magnitude of Ca2+ influx had much smaller influence on [Ca2+]i with a 50% increase of Ca2+ influx resulting in only a 15% increase of peak Ca2+. According to Hilgemann this discrepancy was the result of the different impact of these interventions on the thermodynamics of NCX. While extracellular Ca2+ is also a simple determinant of the exchanger equilibrium, isolated changes of Ca2+ influx would primarily be buffered by the exchanger over several beats [74]. Hence, the observation that myocytes from knock-out mice have unchanged Ca2+ dynamics is supported by the idea of Hilgemann that in murine models NCX is mainly working as a Ca2+ buffer with little influence on the E–C cycle under steady-state conditions.
In our model [73] we found that under voltage clamp conditions the Ca2+ current in myocytes from knock-out mice was reduced by 50% compared to controls although the number of channels remained unchanged (Fig. 3). From these data we hypothesized that gain is increased, in contrast to the reduction in the gain of E–C coupling reported in the transgenic mice overexpressing NCX [48]. By analogy to results from these transgenic mice it would be a speculative possibility that the lack of NCX as Ca2+ buffer in the knock-out would lead to an increase of Ca2+ levels in the diadic cleft promoting Ca2+-dependent inactivation of the L-type Ca2+ current.
4. Conclusion
Cardiac E–C coupling describes the process that links sarcolemmal Ca2+ influx and Ca2+ release from the SR in a subsarcolemmal microdomain of restricted diffusion and high levels of [Ca2+]i called the diadic cleft. NCX has been modeled to be outside of this compartment and is likely co-localized with the Na+-pump in a larger microdomain or ‘fuzzy’ space with increased levels of [Na+]i, which may not strictly coincide but could be adjacent to the diadic cleft. These microdomains with local behavior of ion activity are central to the understanding of NCX in the course of an E–C cycle since [Na+] and [Ca2+] set the thermodynamic equilibrium of the exchanger and directly regulate its activity. Under experimental conditions it could be shown that NCX could directly modulate SR Ca2+ load, SR Ca2+ release as well as Ca2+ spark frequency. Some of these findings could be reproduced in a transgenic mouse model overexpressing the cardiac exchanger. Overexpression by the factor 3.1 resulted in reduced E–C coupling gain and altered behavior of the L-type Ca2+ current with an increased peak Ca2+ influx and slowed inactivation kinetics. These alterations could be explained by a spatial rearrangement of NCX in the plasma membrane at higher densities. With closer proximity to the L-type Ca2+ channel the exchanger could act as a Ca2+ sink or buffer reducing local Ca2+ in the diadic cleft and in the vicinity of the dihydropyridine receptor.
In some respect, the opposite was observed in adult ventricular myocytes from mice with conditional knock-out of the exchanger. These myocytes adapted surprisingly well to the absence of NCX by reducing transsarcolemmal fluxes of Ca2+ and increasing E–C coupling gain. This again is likely associated with changes in submembrane Ca2+ levels and autoregulatory mechanisms that we do not yet fully understand.
References
Author notes
Time for primary review 21 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}