





Abstract
Objective: We tested the hypothesis that simultaneous inhibition of the endothelial integrin αvβ3 and the platelet glycoprotein IIb/IIIa receptor will substantially reduce infarct size in a model of acute coronary thrombosis and primary angioplasty.
Methods: Dogs were subjected to thrombus formation in the left anterior descending coronary artery followed by primary angioplasty. Prior to angioplasty, they were randomized into 3 treatment groups. Group 1 (n=7) received saline; Group 2 (n=9) received MK-383 that inhibits only IIb/IIIa; and Group 3 (n=9) received CP-4715, that inhibits both IIb/IIIa and αvβ3.
Results: There was a 59% reduction in infarct size in dogs receiving CP-4715 compared to controls (p=0.002) and a 37% reduction compared to the dogs receiving MK-383 (p=0.04). Myocardium microthrombi were seen to be reduced similarly with both drugs on post-mortem 99mTc-DMP444 autoradiography that reflects in vivo IIb/IIIa receptor activity. In vivo imaging using echistatin-conjugated and leukocyte-targeted microbubbles revealed significant αvβ3 inhibition and reduction in active leukocyte recruitment only in Group 3 dogs. Myocardial blood flow and regional function after reperfusion were also significantly better in this group.
Conclusion: Simultaneous inhibition of IIb/IIIa and αvβ3 causes a marked reduction in infarct size in a model of acute coronary thrombosis and primary PTCA that is associated with reduced myocardial microthrombi and inflammation, as well as improved myocardial blood flow and regional function. These results may have important implications in the treatment of acute coronary syndromes.
1. Introduction
Many attempts have been made to limit infarct size in acute myocardial infarction. These were made initially in models of persistent coronary occlusion and more recently in those of ischemia–reperfusion. These studies utilized some mechanical means, such as snares, to produce coronary occlusion, whereas in the clinical setting coronary occlusion in acute myocardial infarction is most often caused by a thrombus.
We have recently shown that the thrombus burden influences the size of the no-reflow phenomenon as well as infarct size after attempted coronary intervention [1,2]. Disruption of the thrombus during the procedure is associated with microthromboemboli that contribute to both transient ‘no reflow’ as well as permanent muscle damage. It has also been shown that both IIb/IIIa expression and microvascular fibrin formation increase over time after reperfusion leading to leukocyte and platelet entrapment and microvascular obstruction [3]. Therefore, it is possible that the beneficial effects of platelet glycoprotein IIb/IIIa receptor blockade seen in patients [4,5] may in part be due to reduction in microthromboemboli and consequently in infarct size.
Another consequence of ischemia/reperfusion is endothelial activation from oxygen free radicals and tissue factor resulting in the expression of the endothelial integrin αvβ3 that, in turn, causes platelets to adhere to the endothelium [6]. In the presence of microthromboemboli, prothrombin can bind to both IIb/IIIa and αvβ3 resulting in additional thrombus formation. αvβ3 activation is also associated with leukocyte entrapment within the platelet–fibrin mesh as well as monocyte adhesion to the endothelium [7]. These effects could also reduce microvascular perfusion and cause more ischemia [8]. We, therefore, hypothesized that simultaneuous inhibition of both αvβ3 and IIb/IIIa will substantially reduce infarct size by increasing microvascular perfusion in a model of acute coronary thrombosis and reperfusion.
2. Methods
2.1. Experimental design
We used a canine model of acute coronary thrombosis where primary angioplasty was used as a means of achieving reperfusion. Both αvβ3 and IIb/IIIa were blocked by a novel synthetic compound: CP-4715 (Meiji): (2S)-Benzenesulfonyl-amino-3[3-methoxy-4{4-(1,4,5,6-tetrahydro-pyrimidin-2-ylamino)piperidin-1-yl}benzoylamino]propionic acid [9]. This experimental compound also has a weak inhibitory action on the endothelial integrin α5β1. Its IC50 for αvβ3, IIb/IIIa, and α5β1 are 0.19, 0.44, and 250 nM.L−1, respectively [9]. We compared its action with that of MK-383 (Tirofiban HCl, Merck), which predominantly blocks the IIb/IIIa receptor. The IC50 of MK-383 for IIb/IIIa and αvβ3 are 9 and 62000 nM.L−1, respectively [10].
We utilized molecular imaging to assess inhibition of αvβ3 and IIb/IIIa within the myocardium. For the former, we used in-vivo ultrasound imaging of microbubbles with echistatin incorporated on their surface [11,12], while for the latter we used post-mortem imaging of a 99mTc-labeled molecule (DMP-444) that binds to the IIb/IIIa receptor [13]. We assessed the spatial extent of myocardial inflammation in-vivo with ultrasound imaging of leukocyte-targeted microbubbles [14,15]. We also measured the effect of the drugs on platelet aggregation and bleeding time [16,17].
To determine the effects of inhibiting inflammation and microthrombi on microvascular perfusion, regional myocardial blood flow was measured post-mortem using radiolabeled microspheres [18,19]. This measurement was performed in the central 50% of the risk area in order to eliminate the effect of collateral myocardial blood flow on infarct size. Risk area and the size of the ‘no reflow’ zone were defined using in-vivo contrast echocardiography [18,19]; wall thickening was measured with in-vivo two-dimensional echocardiography [20]; and, infarct size was measured by post-mortem tissue staining with 2.3.5-triphenyl tetrazolium chloride [21].
2.2. Animal preparation
Twenty-five anesthetized open-chest dogs were used for this study that was approved by the Animal Research Committee at the University of Virginia and conformed to the “Guide for the Care and Use of Laboratory Animals” published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). An 8F introducer sheath was placed in the left carotid artery. Catheters were placed in both femoral arteries for duplicate reference sample withdrawal during radiolabeled microsphere injection, and one of them was also connected to a multichannel recorder for arterial pressure monitoring.
Catheters were placed in both femoral veins for administration of fluids and infusion of microbubbles. A heated mattress was used to maintain the core body temperature at 38 °C. A left lateral thoracotomy was performed and the heart was suspended in a pericardial cradle. A 6F catheter was placed in the left atrium for injection of radiolabeled microspheres. The mid and distal portions of the left anterior descending coronary artery as well as its first diagonal branch were dissected free from surrounding tissues.
2.3. Myocardial contrast echocardiography
Intermittent high mechanical index (1.0) power-Doppler harmonic imaging was performed using a Power Vision 6000 system (Toshiba Medical). The transducer was placed in a fixed position in order to obtain the same short-axis view in all stages. A warm water bath acted as an acoustic interface between the heart and the transducer. A dynamic range of 60 dB was employed and color gain was optimized to minimize blooming artifacts during microbubble administration. Wall filter and pulse repetition frequency were adjusted to minimize motion-induced artifacts. Image depth, focus, and gray-scale gain were optimized at the beginning of each experiment and then held constant throughout.
A dose of 0.05 mL of Definity (Bristol Myers Squibb Medical Imaging) was injected intravenously over 30 s followed by a 5 mL saline flush over 1 min. Up to 6 end-systolic images were acquired at a pulsing interval of 8 cardiac cycles before and after contrast administration. The images were converted into gray scale, aligned and averaged, after which the averaged contrast-enhanced image was digitally subtracted from the averaged pre-contrast image. The resultant image was then color-coded and the perfusion defect size (defined as the region without color that included the risk area during coronary occlusion and ‘no reflow’ zones after angioplasty) was planimetered [18,19].
2.4. 99mTc DMP-444 Autoradiography
1 mCi.kg−1 of 99mTc-labeled DMP-444 (Bristol Myers Squibb Medical Imaging) was injected intravenously 180 min after angioplasty. This agent binds to the IIb/IIIa receptor, allowing imaging of the embolized and/or in-situ activated platelets [13]. Its IC50 for the IIb/IIIa receptor is 11 nM.L−1. The spatial extent of myocardial microvascular platelet aggregation was quantified on post-mortem 99mTc-autoradiography, where the heart slice corresponding to the echocardiographic image was imaged directly on the scan-head of a gamma camera (Siemens Orbiter 37) with a high-resolution collimator [2]. After background-subtraction, regions-of-interest were drawn over the risk area as well as the normal posterior wall and a count ratio per pixel between the two regions was computed.
2.5. Microbubbles targeted to leukocyte and αv integrins
Lipid microbubbles targeted to activated leukocytes were prepared by sonication of decafluorobutane gas with an aqueous dispersion of polyethyleneglycol stearate, distearoyl phosphatidylcholine, and distearoyl phosphatidylserine (Avanti Polar Lipids) [14,15]. The in-vivo backscatter from these bubbles is related to both tissue leukocyte count as well as myeloperoxidase activity both in the kidney and heart [14,15]. For microbubbles targeted to αvβ3, biotinylated lipid-shelled microbubbles containing decafluorobutane gas were prepared and 2.5 μg of biotinylated echistatin (Sigma) was conjugated to their surface [12]. The degree of backscatter from these αvβ3-targeted microbubbles is related to in-vivo αvβ3 activity [22]. Vascular endothelial αvβ3 receptor activation has recently been reported early in a canine model of ischemia–reperfusion [23].
For myocardial imaging, 1.108 targeted microbubbles were injected as an intravenous bolus and imaging was initiated 15 min later in order to allow microbubble adhesion to their targets and clearance of freely circulating microbubbles from the blood pool. The first frame was acquired that reflects the total concentration of microbubbles in tissue (both retained and freely circulating). Microbubbles within the beam were then destroyed by continuous imaging at high acoustic power after which the first image reflecting only freely circulating microbubbles was acquired. This image was subtracted from the previously acquired image in order to obtain a single color-coded image representing retained bubbles alone [15]. Backscatter was measured from regions-of-interest placed over the risk area.
2.6. Experimental protocol
Heart rate and mean aortic pressure were recorded throughout the experiment. A bolus injection of 50 U.kg−1 of heparin was administered intravenously. A guide wire was inserted into the left anterior descending coronary artery and a balloon (3.9 mm) was advanced over it to the proximal portion of the artery. Umbilical tapes were tightened around the distal portion of the artery as well as the origin of the first diagonal branch. The mid portion of the left anterior descending coronary artery was subjected to external injury using forceps. Balloon inflation was then performed for 100 min at 6–8 atmospheric pressure in order to isolate the left anterior descending artery segment between the balloon and the distal occlusion site and allow a thrombus to form within it. Heparin was administered very slowly (0.1 unit.mL−1 of saline.kg−1.min−1) via the angioplasty catheter in order to prevent clots from occurring within the catheter itself (Fig. 1).
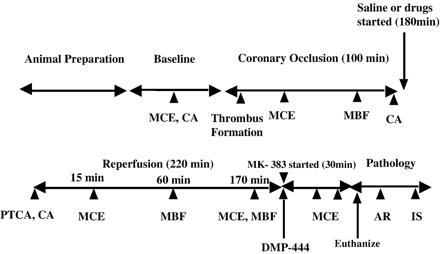
Schema of the experimental protocol. MCE=myocardial contrast echocardiography; CA=coronary angiography, MBF=myocardial blood flow measurement (using radiolabeled microspheres); PTCA=percutaneous transluminal coronary angioplasty; AR=autoradiography (using 99mTc); and IS=infarct size measurement (using triphenyl tetrazolium chloride).
Ten minutes before recanalization, the dogs were randomly allocated to receive either saline (Group 1, 7 dogs); MK-383 (120 μg.kg−1 bolus followed by a continuous infusion of 260 μg.kg−1.h−1 for 3 h–Group 2, 9 dogs); or CP-4715 (15 μg.kg−1 bolus followed by a continuous infusion of 10μg.kg−1.h−1 for 3 h–Group 3, 9 dogs). The distal left anterior descending coronary artery and the first diagonal branch ligations were reversed and the angioplasty balloon was deflated. Coronary angiography was performed to demonstrate the presence of an occlusive thrombus, after which angioplasty was performed and Thrombolysis in Myocardial Infarction grade 3 flow was confirmed on repeat coronary angiography.
Regional wall thickening [20], myocardial contrast echocardiography [18,19], and radiolabeled microsphere-derived myocardial blood flow [18,19] were measured at baseline, during coronary occlusion, and after reperfusion. During reperfusion, myocardial contrast echocardiography was performed after an intravenous bolus injection of 10 mg of adenosine to define the size of the ‘no reflow’ zone. This maneuver accurately defines the in vivo infarct size and prevents hyperemia-induced underestimation of no-reflow [18,19]. All drugs were discontinued immediately after DMP-444 injection.
Myocardial contrast echocardiography was repeated using microbubbles conjugated to echistatin for measuring the degree of αvβ3 activity [12]. Because echistatin also has some affinity for the IIb/IIIa receptor (IC50 of 70 nM), all dogs received a 240 μg.kg−1 bolus injection of MK-383 followed by a continuous infusion of 520 μg.kg−1.h−1 until the end of experiment in order to inhibit any residual IIb/IIIa receptor. Thus, any signal from echistatin conjugated microbubbles was a result of αvβ3 binding and not from binding to IIb/IIIa. After continuous high mechanical index imaging in order to destroy any adherent echistatin conjugated microbubbles, leukocyte-targeted microbubbles were injected to delineate the spatial extent of inflammation (195 min after recanalization) [14]. Sutures were placed on the anterior myocardium at the site of the imaging plane. At 220 min after recanalization, the dogs were euthanized, and a 1 cm thick slice was obtained from the suture site for 99mTc-autoradiography as well as myocardial blood flow and infarct size determinations.
2.7. Statistical methods
Data are expressed as mean ± 1S.D. Data within groups were compared with ANOVA with inter-group comparisons performed using Scheffe's post-hoc test. In order to test for differences in the relation between the infarct size/risk area ratio and these variables, ANCOVA was performed using the infarct size/risk area ratio as the dependent variable [24]. A p value of <0.05 (two-sided) was considered significant.
3. Results
Table 1 presents a summary of the hemodynamic and imaging results. The coronary occlusion time was similar for all 3 groups of dogs. Heart rate, mean aortic pressure, and rate-pressure product were similar for the same stages between all 3 groups of dogs.
Hemodynamic and imaging results (mean ± 1S.D.)
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Coronary occlusion time (min) (range) | 114 ± 4 (110–118) | 113 ± 8 (107–124) | 112 ± 8 (108–119) | 0.76 | |
| Heart rate (beats.min−1) | Baseline | 112 ± 19 | 101 ± 15 | 99 ± 17 | 0.30 |
| Occlusion | 91 ± 9 | 93 ± 8 | 80 ± 17 | 0.14 | |
| Reperfusion | 77 ± 18 | 71 ± 8 | 63 ± 13 | 0.14 | |
| Mean aortic pressure (mm Hg) | Baseline | 74 ± 27 | 89 ± 22 | 87 ± 22 | 0.22 |
| Occlusion | 66 ± 24 | 72 ± 25 | 68 ± 22 | 0.89 | |
| Reperfusion | 60 ± 15 | 57 ± 21 | 60 ± 12 | 0.91 | |
| RA (% of LV myocardium) | 43 ± 11 | 41 ± 12 | 44 ± 7 | 0.64 | |
| No-reflow (% of RA) | 15 mina | 45 ± 27 | 25 ± 14 | 22 ± 16b | 0.05 |
| 170 mina | 35 ± 29 | 24 ± 27 | 6 ± 5b,c,d | 0.04 | |
| % Wall thickening | Baseline | 31 ± 10 | 33 ± 11 | 35 ± 9 | 0.94 |
| Occlusion | 8 ± 8 | 7 ± 3 | 7 ± 6 | 0.97 | |
| Reperfusion | 12 ± 7 | 14 ± 8 | 24 ± 9b,c | 0.02 | |
| 99mTc-DMP444 activity (normalized) | 2.7 ± 1.0 | 1.7 ± 0.5b | 1.6 ± 0.3b | 0.004 | |
| Backscatter from Echistatin- conjugated micobubbles | 111 ± 20 | 118 ± 26 | 80 ± 21b,c | 0.04 | |
| Backscatter from Leukocyte- targeted microbubbles | 125 ± 27 | 120 ± 22 | 87 ± 18b | 0.05 | |
| IS (% of LV myocardium) | 24 ± 2 | 14 ± 7b | 11 ± 6b | 0.0004 | |
| IS/RA ratio | 59 ± 13 | 37 ± 15c | 24 ± 12b,c | 0.0002 |
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Coronary occlusion time (min) (range) | 114 ± 4 (110–118) | 113 ± 8 (107–124) | 112 ± 8 (108–119) | 0.76 | |
| Heart rate (beats.min−1) | Baseline | 112 ± 19 | 101 ± 15 | 99 ± 17 | 0.30 |
| Occlusion | 91 ± 9 | 93 ± 8 | 80 ± 17 | 0.14 | |
| Reperfusion | 77 ± 18 | 71 ± 8 | 63 ± 13 | 0.14 | |
| Mean aortic pressure (mm Hg) | Baseline | 74 ± 27 | 89 ± 22 | 87 ± 22 | 0.22 |
| Occlusion | 66 ± 24 | 72 ± 25 | 68 ± 22 | 0.89 | |
| Reperfusion | 60 ± 15 | 57 ± 21 | 60 ± 12 | 0.91 | |
| RA (% of LV myocardium) | 43 ± 11 | 41 ± 12 | 44 ± 7 | 0.64 | |
| No-reflow (% of RA) | 15 mina | 45 ± 27 | 25 ± 14 | 22 ± 16b | 0.05 |
| 170 mina | 35 ± 29 | 24 ± 27 | 6 ± 5b,c,d | 0.04 | |
| % Wall thickening | Baseline | 31 ± 10 | 33 ± 11 | 35 ± 9 | 0.94 |
| Occlusion | 8 ± 8 | 7 ± 3 | 7 ± 6 | 0.97 | |
| Reperfusion | 12 ± 7 | 14 ± 8 | 24 ± 9b,c | 0.02 | |
| 99mTc-DMP444 activity (normalized) | 2.7 ± 1.0 | 1.7 ± 0.5b | 1.6 ± 0.3b | 0.004 | |
| Backscatter from Echistatin- conjugated micobubbles | 111 ± 20 | 118 ± 26 | 80 ± 21b,c | 0.04 | |
| Backscatter from Leukocyte- targeted microbubbles | 125 ± 27 | 120 ± 22 | 87 ± 18b | 0.05 | |
| IS (% of LV myocardium) | 24 ± 2 | 14 ± 7b | 11 ± 6b | 0.0004 | |
| IS/RA ratio | 59 ± 13 | 37 ± 15c | 24 ± 12b,c | 0.0002 |
RA=risk area; IS=infarct size; LV=left ventricular.
After reperfusion.
p<0.05 vs. Group 1.
p<0.05 vs. Group 2.
Compared to 15 min after reperfusion.
Hemodynamic and imaging results (mean ± 1S.D.)
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Coronary occlusion time (min) (range) | 114 ± 4 (110–118) | 113 ± 8 (107–124) | 112 ± 8 (108–119) | 0.76 | |
| Heart rate (beats.min−1) | Baseline | 112 ± 19 | 101 ± 15 | 99 ± 17 | 0.30 |
| Occlusion | 91 ± 9 | 93 ± 8 | 80 ± 17 | 0.14 | |
| Reperfusion | 77 ± 18 | 71 ± 8 | 63 ± 13 | 0.14 | |
| Mean aortic pressure (mm Hg) | Baseline | 74 ± 27 | 89 ± 22 | 87 ± 22 | 0.22 |
| Occlusion | 66 ± 24 | 72 ± 25 | 68 ± 22 | 0.89 | |
| Reperfusion | 60 ± 15 | 57 ± 21 | 60 ± 12 | 0.91 | |
| RA (% of LV myocardium) | 43 ± 11 | 41 ± 12 | 44 ± 7 | 0.64 | |
| No-reflow (% of RA) | 15 mina | 45 ± 27 | 25 ± 14 | 22 ± 16b | 0.05 |
| 170 mina | 35 ± 29 | 24 ± 27 | 6 ± 5b,c,d | 0.04 | |
| % Wall thickening | Baseline | 31 ± 10 | 33 ± 11 | 35 ± 9 | 0.94 |
| Occlusion | 8 ± 8 | 7 ± 3 | 7 ± 6 | 0.97 | |
| Reperfusion | 12 ± 7 | 14 ± 8 | 24 ± 9b,c | 0.02 | |
| 99mTc-DMP444 activity (normalized) | 2.7 ± 1.0 | 1.7 ± 0.5b | 1.6 ± 0.3b | 0.004 | |
| Backscatter from Echistatin- conjugated micobubbles | 111 ± 20 | 118 ± 26 | 80 ± 21b,c | 0.04 | |
| Backscatter from Leukocyte- targeted microbubbles | 125 ± 27 | 120 ± 22 | 87 ± 18b | 0.05 | |
| IS (% of LV myocardium) | 24 ± 2 | 14 ± 7b | 11 ± 6b | 0.0004 | |
| IS/RA ratio | 59 ± 13 | 37 ± 15c | 24 ± 12b,c | 0.0002 |
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Coronary occlusion time (min) (range) | 114 ± 4 (110–118) | 113 ± 8 (107–124) | 112 ± 8 (108–119) | 0.76 | |
| Heart rate (beats.min−1) | Baseline | 112 ± 19 | 101 ± 15 | 99 ± 17 | 0.30 |
| Occlusion | 91 ± 9 | 93 ± 8 | 80 ± 17 | 0.14 | |
| Reperfusion | 77 ± 18 | 71 ± 8 | 63 ± 13 | 0.14 | |
| Mean aortic pressure (mm Hg) | Baseline | 74 ± 27 | 89 ± 22 | 87 ± 22 | 0.22 |
| Occlusion | 66 ± 24 | 72 ± 25 | 68 ± 22 | 0.89 | |
| Reperfusion | 60 ± 15 | 57 ± 21 | 60 ± 12 | 0.91 | |
| RA (% of LV myocardium) | 43 ± 11 | 41 ± 12 | 44 ± 7 | 0.64 | |
| No-reflow (% of RA) | 15 mina | 45 ± 27 | 25 ± 14 | 22 ± 16b | 0.05 |
| 170 mina | 35 ± 29 | 24 ± 27 | 6 ± 5b,c,d | 0.04 | |
| % Wall thickening | Baseline | 31 ± 10 | 33 ± 11 | 35 ± 9 | 0.94 |
| Occlusion | 8 ± 8 | 7 ± 3 | 7 ± 6 | 0.97 | |
| Reperfusion | 12 ± 7 | 14 ± 8 | 24 ± 9b,c | 0.02 | |
| 99mTc-DMP444 activity (normalized) | 2.7 ± 1.0 | 1.7 ± 0.5b | 1.6 ± 0.3b | 0.004 | |
| Backscatter from Echistatin- conjugated micobubbles | 111 ± 20 | 118 ± 26 | 80 ± 21b,c | 0.04 | |
| Backscatter from Leukocyte- targeted microbubbles | 125 ± 27 | 120 ± 22 | 87 ± 18b | 0.05 | |
| IS (% of LV myocardium) | 24 ± 2 | 14 ± 7b | 11 ± 6b | 0.0004 | |
| IS/RA ratio | 59 ± 13 | 37 ± 15c | 24 ± 12b,c | 0.0002 |
RA=risk area; IS=infarct size; LV=left ventricular.
After reperfusion.
p<0.05 vs. Group 1.
p<0.05 vs. Group 2.
Compared to 15 min after reperfusion.
3.1. No-reflow size
While the risk area size was similar between the 3 groups, the ‘no reflow’ zone was different between all 3 groups at both 15 and 170 min after reperfusion. Fig. 2 shows the ‘no reflow’ zone size in a single dog from each of the 3 groups 15 min after recanalization. It was the largest (47% of risk area) in the Group 1 dog receiving saline (panel A), intermediate (38% of risk area) in the Group 2 dog receiving MK-383 (panel B), and smallest (18% of the risk area) in the Group 3 dog receiving CP-4715. The size of the no reflow zone decreased between 15 and 170 min only in Group 3 dogs (Table 1), possibly by inhibiting leukocyte migration (see later). While percent wall thickening was similar in all 3 groups at baseline, it was significantly higher in the Group 3 dogs at 165 min after reperfusion compared to controls (Table 1).
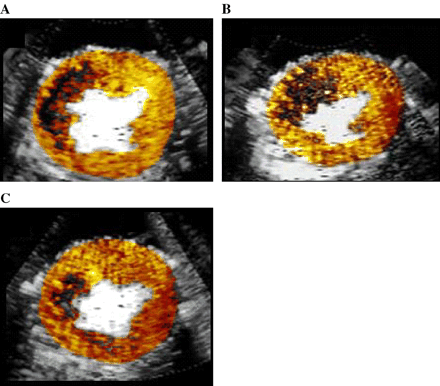
Myocardial contrast echocardiography-derived ‘no reflow’ zones for images from dogs receiving A) saline (Group 1), B) MK-383 (Group 2), and CP-4715 (Group 3).
3.2. Myocardial IIb/IIIa activity
99mTc-DMP444 activity on autoradiography, reflecting IIb/IIIa receptors on myocardial microvascular platelets, was similar in the Groups 2 and 3 dogs, which was significantly lower than in the Group 1 dogs at 180 min after reperfusion (Table 1). Fig. 3 illustrates 99mTc-DMP444 images from the same dogs as in Fig. 2. The activity ratio was highest (3.51) in the Group 1 dog receiving saline (panel A) compared to the Group 2 dog (1.71) receiving MK-383 (panel B) or the Group 3 dog (1.42) receiving CP-4715 (panel C). These results indicate substantial inhibition of the IIb/IIIa receptor by CP-4715 and MK-383.
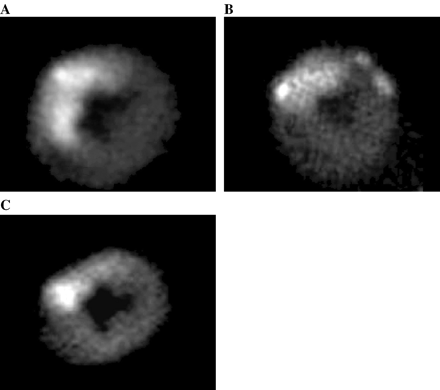
Platelet aggregation in the myocardium 180 min after recannalization on 99mTc-DMP-444 autoradiography. A), B) and C) same as in Fig. 2.
3.3. Myocardial αvβ3 activity
Myocardial backscatter from the echistatin-conjugated microbubbles on MCE was the lowest in Group 3 dogs and similar in the other 2 groups (Table 1). Fig. 4 shows backscatter from echistatin-conjugated microbubbles in the same dogs shown in Figs. 2 and 3. It was high (138) in the Group 1 dog receiving saline (panel A) and Group 2 dog (115) receiving MK-383 (panel B), but was significantly less in the Group 3 dog (32) receiving CP-4715 (panel C). These results indicate marked suppression of αvβ3 by CP-4715.
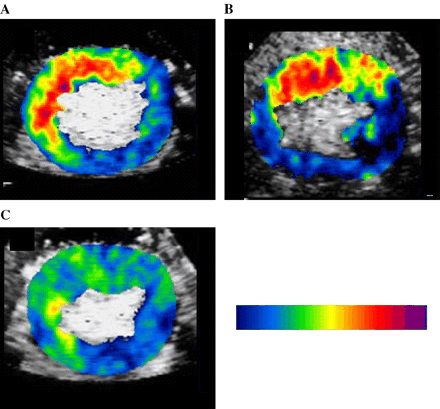
Myocardial αvβ3 activity measured using backscatter from echistatin-conjugated microbubbles on 99mTc-autoradiography A), B) and C) same as in Fig. 2.
3.4. Myocardial inflammation
Fig. 5 shows myocardial backscatter from the leukocyte-targeted microbubbles from the same dogs shown in Figs. 2–4. This backscatter was high in the Group 1 dog (135) receiving saline (panel A) and the Group 2 dog (114) receiving MK-383 (panel B), whereas it was significantly lower in the Group 3 dog (42) receiving CP-4715 (panel C), indicating suppression of active leukocyte recruitment by CP-4715. Similar results were evident in all dogs Table 1.
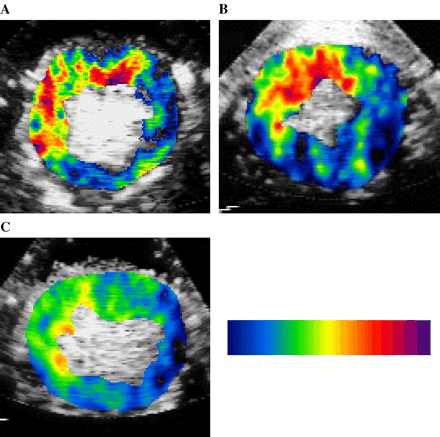
‘Inflammation’ imaging from phopstadylserine-conjugated microbubbles. A), B) and C) same as in Fig. 2.
3.5. Regional myocardial blood flow
Table 2 shows transmural myocardial blood flow within the central 50% of the risk area during the different stages. While myocardial blood flow during coronary occlusion was similar for all 3 groups, the infarct size/risk area ratio was significantly different (Table 1). Multivariate regression analysis revealed that pharmacological intervention was the only independent variable that correlated with the infarct size/risk area ratio (F=25.7, p<0.0001). At both 1 and 3 h after reperfusion, myocardial blood flow was significantly higher in the treated compared to the control dogs indicating that the drug treatment had a beneficial effect on myocardial blood flow after reperfusion probably by inhibiting platelet aggregation and leukocyte trafficking within the microcirculation. Although not statistically significant, the Group 3 dogs tended to have the highest myocardial blood flow at 3 h after reperfusion. A strong inverse relation was noted between myocardial blood flow after reperfusion and the infarct size/risk area ratio in this group of dogs: y=−016x+0.47 (r=0.74, p=0.02).
Myocardial blood flow data (mean ± 1S.D.) (mL.min−1.g−1) within the central 50% of the risk area
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| During coronary occlusion | 0.07 ± 0.05 | 0.11 ± 0.04 | 0.10 ± 0.05 | 0.33 | |
| After reperfusion | 60 min | 0.68 ± 0.22 | 1.04 ± 0.28a | 1.09 ± 0.09a | 0.002 |
| 180 min | 0.58 ± 0.17 | 0.87 ± 0.19a | 1.02 ± 0.17a | <0.001 |
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| During coronary occlusion | 0.07 ± 0.05 | 0.11 ± 0.04 | 0.10 ± 0.05 | 0.33 | |
| After reperfusion | 60 min | 0.68 ± 0.22 | 1.04 ± 0.28a | 1.09 ± 0.09a | 0.002 |
| 180 min | 0.58 ± 0.17 | 0.87 ± 0.19a | 1.02 ± 0.17a | <0.001 |
p<0.05 vs. Group 1.
Myocardial blood flow data (mean ± 1S.D.) (mL.min−1.g−1) within the central 50% of the risk area
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| During coronary occlusion | 0.07 ± 0.05 | 0.11 ± 0.04 | 0.10 ± 0.05 | 0.33 | |
| After reperfusion | 60 min | 0.68 ± 0.22 | 1.04 ± 0.28a | 1.09 ± 0.09a | 0.002 |
| 180 min | 0.58 ± 0.17 | 0.87 ± 0.19a | 1.02 ± 0.17a | <0.001 |
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| During coronary occlusion | 0.07 ± 0.05 | 0.11 ± 0.04 | 0.10 ± 0.05 | 0.33 | |
| After reperfusion | 60 min | 0.68 ± 0.22 | 1.04 ± 0.28a | 1.09 ± 0.09a | 0.002 |
| 180 min | 0.58 ± 0.17 | 0.87 ± 0.19a | 1.02 ± 0.17a | <0.001 |
p<0.05 vs. Group 1.
3.6. Infarct size
Despite the same sized risk area, the infarct size was different between the 3 groups (Table 1). Fig. 6 shows the final infarct size in the same dogs shown in Figs. 2–5. It was the largest in the Group 1 dog (45% of risk area) receiving saline (panel A), intermediate in the Group 2 dog (34% of risk area) receiving MK-383 (panel B), and smallest in the Group 3 dog (18% of the risk area) receiving CP-4715. Overall, there was a 59% reduction in infarct size in the Group 3 dogs receiving CP-4715 compared to controls (p=0.002) and a 37% reduction compared to the group 2 dogs receiving MK-383 (p=0.04). The infarct size was significantly different (p=0.03) between the 3 groups even after normalizing for myocardial blood flow during coronary occlusion.
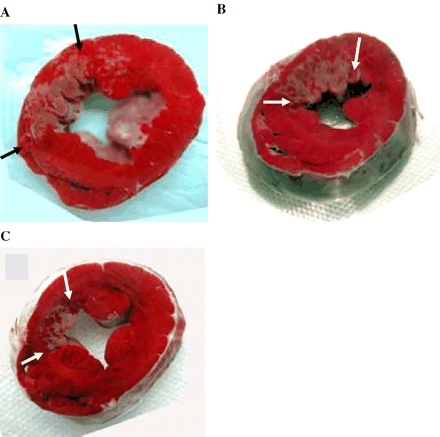
Post-mortem triphenyl tetrazolium chloride-defined infarct size (arrows). A), B), and C) same as in Fig. 2.
3.7. Other variables
Table 3 summarizes the results of ex-vivo platelet aggregation and bleeding time measurements at 60 and 180 min after reperfusion and prior to the administration of MK-383 in all dogs. While platelet aggregation was inhibited at different points in time in both Group 2 and 3 dogs, bleeding time was significantly prolonged only in Group 2 dogs.
Platelet aggregation and bleeding times (mean ± 1S.D.)
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Inhibition of platelet aggregation (% of baseline) | 60 min | −6 ± 17a | 93 ± 12 | 86 ± 7 | 0.0002 |
| 180 min | −2 ± 14a | 96 ± 4 | 85 ± 7 | 0.0001 | |
| Bleeding time (ratio of baseline) | 60 min | 1.0 ± 0.1 | 1.8 ± 0.1a | 1.1 ± 0.3 | 0.003 |
| 180 min | 0.9 ± 0.2 | 1.7 ± 0.2a | 1.0 ± 0.3 | 0.004 |
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Inhibition of platelet aggregation (% of baseline) | 60 min | −6 ± 17a | 93 ± 12 | 86 ± 7 | 0.0002 |
| 180 min | −2 ± 14a | 96 ± 4 | 85 ± 7 | 0.0001 | |
| Bleeding time (ratio of baseline) | 60 min | 1.0 ± 0.1 | 1.8 ± 0.1a | 1.1 ± 0.3 | 0.003 |
| 180 min | 0.9 ± 0.2 | 1.7 ± 0.2a | 1.0 ± 0.3 | 0.004 |
p<0.05 vs. other groups.
Platelet aggregation and bleeding times (mean ± 1S.D.)
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Inhibition of platelet aggregation (% of baseline) | 60 min | −6 ± 17a | 93 ± 12 | 86 ± 7 | 0.0002 |
| 180 min | −2 ± 14a | 96 ± 4 | 85 ± 7 | 0.0001 | |
| Bleeding time (ratio of baseline) | 60 min | 1.0 ± 0.1 | 1.8 ± 0.1a | 1.1 ± 0.3 | 0.003 |
| 180 min | 0.9 ± 0.2 | 1.7 ± 0.2a | 1.0 ± 0.3 | 0.004 |
| Variable | Group 1 Saline (n=7) | Group 2 MK-383 (n=9) | Group 3 CP-4715 (n=9) | P value | |
|---|---|---|---|---|---|
| Inhibition of platelet aggregation (% of baseline) | 60 min | −6 ± 17a | 93 ± 12 | 86 ± 7 | 0.0002 |
| 180 min | −2 ± 14a | 96 ± 4 | 85 ± 7 | 0.0001 | |
| Bleeding time (ratio of baseline) | 60 min | 1.0 ± 0.1 | 1.8 ± 0.1a | 1.1 ± 0.3 | 0.003 |
| 180 min | 0.9 ± 0.2 | 1.7 ± 0.2a | 1.0 ± 0.3 | 0.004 |
p<0.05 vs. other groups.
4. Discussion
The new information from this study is that simultaneous inhibition of the platelet IIb/IIIa receptor and the endothelial integrin αvβ3 causes a marked reduction in infarct size in a model of acute coronary thrombosis and primary angioplasty. Myocardial perfusion is better in regions where myocardial inflammation and microthrombi are reduced after reperfusion, potentially leading to less necrosis. These results may also explain, in part, the beneficial results seen with IIb/IIIa inhibition in patients with acute coronary syndromes [4,5] and suggest that combining IIb/IIIa receptor inhibition with concomitant antagonism of αvβ3 could be very useful in such patients, particularly when undergoing coronary interventions.
4.1. Inhibition of the platelet IIb/IIIa receptor
Two studies that evaluated the effect of IIb/IIIa inhibition on infarct size reported diametrically opposite results [25,26], which may in part be related to the model used–that of acute coronary ligation without significant thrombus production. Although IIb/IIIa receptor activation can occur within the microvasculature during myocardial ischemia, the degree of activation is mild and so in-situ microthrombus formation is minimal [2]. On the other hand, when there is substantial microthromboembolism resulting from angioplasty performed for acute coronary thrombosis, IIb/IIIa receptor activation is markedly increased [1,2]. Thus, positive results from IIb/IIIa inhibition in this situation are more likely than during acute coronary ligation. In this study, we noted a marked reduction in myocardial microthrombi with MK-383 and CP-4715 as evidenced by reduced myocardial 99mTc-labeled DMP444 activity.
In a recent study of patients with acute coronary syndromes, aspiration of the coronary artery showed thrombus with or without plaque components in half the patients. The other half had only plaque consisting of cholesterol crystals [27]. It is likely that in the latter release of tissue factor and other substances also activated the platelet IIb/IIIa receptors, resulting in their aggregation and in-situ microthrombus formation [6]. Thus, the beneficial effects of IIb/IIIa inhibition in acute coronary syndromes may also result from infarct size reduction by prevention of in-situ microthrombus formation that could explain the better results seen in patients with acute coronary syndromes when they undergo x-sizer laser thrombectomy prior to angioplasty and/or stenting [28].
4.2. Inhibition of the endothelial integrin αvβ3
There have been no studies so far that have demonstrated a reduction in infarct size with an αvβ3 antagonist in a model of acute coronary thrombosis. This integrin is expressed on both sides of the endothelial cell surface. On the outside it anchors the endothelial cells to smooth muscle and the intercellular matrix, thus maintaining capillary hydraulic conductivity [29]. When activated on the luminal surface, it binds to several plasma proteins such as fibrinogen, vitronectin, thrombospondin, and prothrombin [6,8]. It also causes endothelial adhesion of activated platelets via fibrinogen linkage [6], and is thought to participate in the inflammatory process by the entrapment of leukocytes within the platelet–fibrin mesh as well as by direct adhesion of monocytes on endothelial cells [7]. Thus, αvβ3 expression upregulation can cause microvascular obstruction via several mechanisms. It is also coexpressed in ischemia along with matrix mettaloproteinases that participate in tissue lysis during infarction [30]. This upregulation takes only a few hours to occur and is predominantly seen in small (30–50 μm sized) arterioles [3]. Our results indicate that this upregulation is inhibited by CP-4715 as evidenced by reduced myocardial retention of echistatin-conjugated microbubbles.
The potentiating effect of αvβ3 antagonism on IIb/IIIa receptor inhibition on infarct size reduction indicates that αvβ3 antagonism results in beneficial effects from mechanisms other than just inhibition of platelet–endothelial interactions. The marked reduction in myocardial inflammation with CP-4715 seen in our study suggests that leukocyte entrapment within the microcirculation may be reduced, leading to less no-reflow, better perfusion, smaller infarct size, less edema, and better regional function post-reperfusion. Our results imply that these benefits are achieved by reducing reperfusion-induced myocardial injury.
4.3. Critique of our methods
This was a complicated study requiring many measurements. However, the unique combination of pharmacology, in-vivo molecular imaging, and post-mortem tissue analysis is a strength of this study. In order to abolish any cross-talk from any residual IIb/IIIa receptor during imaging of αvβ3 with echistatin-conjugated microbubbles, we administered MK-383 in all the dogs at the end of the experiment. This maneuver may have resulted in some reduction in final infarct size: however, all 3 groups received the drug and any potential effect on infarct size should have been similar.
Whereas αvβ3 inhibition seems to be an important factor in limiting infarct size in this study, we did not have a group where this integrin was solely inhibited. Since the major difference between MK-383 and CP-4715 was the inhibition of αvβ3, it is reasonable to assume that most of the benefit of CP-4715 was from inhibition of this integrin. Histologic studies would have been useful in this regard. Consequently, further studies are required to determine the specific role of αvβ3 inhibition alone for reducing infarct size.
We could have used Abciximab instead of CP-4715. The advantages of the latter are its lack of effect on bleeding time at doses required to reduce infarct size as well as its anti-allergic properties because of its non-peptide structure. Platelet aggregation can also play a role in the microvascular obstruction during inflammation, but inhibition of aggregation was equal in both Group 2 and 3 dogs while inflammation was inhibited only in Group 3 dogs.
The backscatter from phosphatidylserine coated microbubbles has been compared to tissue myeloperoxidase activity in a canine model of myocardial ischemia and reperfusion similar to that used in this study [15]. A good correlation was found even up to 120 min after reperfusion. We believe that the results should be similar even at 6 h after reperfusion, when the number of both intravascular and extravasated leukocytes should have peaked. After that the number of intravascular leukocytes that can be detected by the microbubbles should decrease while the number of extravasated leukocytes should still be high. At that point in time the correlation between backscatter from phosphatidylserine coated microbubbles and myeloperoxidase activity may not be that good.
We selected a pulsing interval of 8 cardiac cycles for myocardial contrast echocardiography because in our preliminary studies we found that this interval gave us the best estimation of infarct size during transient exogenous hyperemia and risk area during rest. Using a single interval simplified the protocol. Cross-talk between different targeted microbubbles was not an issue since all bubbles were destroyed with high-mechanical index imaging prior to the next stage.
4.4. Clinical implications
There is now enough clinical evidence that acute coronary syndromes, with or without a coronary intervention, are associated with microvascular thromboembolism and myocardial infarction. Our results indicate that simultaneous inhibition of the platelet IIb/IIIa receptor and the endothelial integrin αvβ3, prior to and for some hours following reperfusion, reduces infarct size by diminishing myocardial microthrombi and inflammation and possibly also improving microvascular perfusion. Clinical trials are needed to determine whether this strategy will be effective in patients.
Acknowledgements
Supported in part by grants from the National Institutes of Health (3R01-HL-48890), Bethesda, Maryland, and Meiji Seika Kaisha, Yokohama, Japan. Dr. Sari was supported by a fellowship training grant from the Turkish Cardiac Society and Dr. Lindner was the recipient of the Mentored Clinical Investigator Award (K08-HL03810) from the National Institutes of Health.
References
Author notes
Presented in part at the 52nd Annual Scientific Session of the American College of Cardiology, April 2003, in Chicago
Time for primary review 15 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}