





Abstract
Scavenger receptors (SRs), which recognize modified low-density lipoprotein (LDL) by oxidation or acetylation, are a group of receptors on plasma membrane of macrophages and other cell types. These receptors by facilitating modified LDL uptake are a primary step in the intracellular accumulation of modified LDL and formation of fatty streak. Non-coding RNAs (ncRNAs) are a group of functional RNA nucleotides that are not translated into protein, and include microRNAs (miRs), snoRNAs, siRNAs, snRNAs, exRNAs, piRNAs, and the long ncRNAs (lncRNAs). Recently, ncRNAs have received much attention due to their effects in a variety of disease states such as atherosclerotic cardiovascular disease and cancers. A host of ncRNAs, such as miRs and lncRNAs, have been found to be involved in the regulation of SRs and the inflammatory cascade and subsequently atherosclerosis. Here, we review this important area to create interest in this growing field among researchers and clinicians alike.
1. Introduction
Scavenger receptors (SRs) participate in the removal of many foreign substances and waste materials by extensive ligand specificity.1 In atherosclerotic lesions, macrophages are very prominent; these cells express several SRs (e.g. SR-A and SR-B and lectin-type-oxidized LDL receptor 1) on their membrane for uptake of oxidized low-density lipoprotein (ox-LDL). These lipid-laden macrophages then transform into foam cells resulting in the formation of fatty streak. These foam cells secrete various inflammatory cytokines and trigger a series of intracellular events, such as activation of NADPH oxidases and redox-sensitive pathways, and accelerate the development of atherosclerosis.2 The strategy of blockade of the SR expression has been widely studied and used in the treatment of disease states such as atherosclerosis.3,4
Human genome has a large sequence, of which <2% are protein-coding sequences. The remaining 98%, named as non-coding RNAs (ncRNA), had been thought of as non-functional or ‘junk’ until 10 years ago. These ncRNAs are not translated into protein, and include microRNAs (miRs), snoRNAs, siRNAs, snRNAs, exRNAs, piRNAs, and the long ncRNAs (lncRNAs). A growing body of work shows that a large part of these ncRNAs are engaged in various physiological and pathological processes such as inflammation, atherosclerosis, and cancers.5,6 As such, these ncRNAs could be considered ‘functional’ instead of ‘junk’.
Here, we define the relationship between SRs and ncRNAs, especially miRs and lncRNAs. A detailed knowledge of the function of ncRNAs may have a diagnostic as well as therapeutic value.
2. Classification of scavenger receptors
SRs are divided into 10 classes according to their structural characteristics (Classes A–J). Macrophages express a large number of SRs. Other cell types also express a variety of SRs although in varying density.
Some SRs are known by multiple names. For example, SCARA1 is also known as SR-AI, CD204, or MSR1, and SCARA3 is also known as MSRL1.
The classification of SRs with proposed nomenclature and chromosomal location in humans is summarized in Table 1.
Category of SRs in human
| Current NCBI gene name | ALT names | Proposed nomenclaturea | Protein molecular weight | Accession no. | NCBI gene ID | CHR | ncRNA |
|---|---|---|---|---|---|---|---|
| SCARA1 | SR-A1 CD204 MSR1 | SR-A1 | 50 kDa | NM_138715 | 4481 | 8 | miR-29a miR-155 miR-125b |
| SCARA3 | MSRL1 | SR-A3 | 65 kDa | NM_016240 | 51435 | 8 | |
| COLEC12 | SCARA4 SRCL CL-P1 | SR-A4 | 127 kDa | NM_130386 | 81035 | 18 | |
| SCARA5 | TESR NET33 | SR-A5 | 43 kDa | NM_173833 | 286133 | 8 | |
| MARCO | SCARA2 | SR-A6 | 53 kDa | NM_006770 | 8685 | 2 | |
| SCARB1 | SR-B1 CD36L1 | SR-B1 | 54 kDa | NM_005505 | 949 | 12 | miR-125a miR-455 miR-96 miR-185 miR-223 |
| SCARB2 | LIMP2 CD36L2 LGP85 | SR-B1.1 | 54 kDa | NM_005506 | 950 | 4 | |
| CD36 | SCARB3 FAT GPIV PAS4 | SR-B2 | 88 kDa | NM_001001548 | 948 | 7 | miR-155 miR-27a/b |
| CD68 | gp110 SCARD1 LAMP4 | SR-D1 | 37 kDa | NM_001251 | 968 | 17 | miR-27a miR-146 miR-328 miR-30ea |
| OLR1 | LOX-1 SCARE1 CLEC8A | SR-E1 | 31 kDa | NM_002543 | 4973 | 12 | let-7g miR-29b miR-369-3p miR-370 |
| Dectin 1 | CLEC7A | SR-E2 | 28 kDa | NM_197947 | 64581 | 12 | |
| SCARF1 | SREC-1 | SR-F1 | 87 kDa | NM_003693 | 8578 | 17 | |
| SCARF2 | SREC-II | SR-F2 | 92 kDa | NM_153334 | 91179 | 22 | |
| MEGF10 | EMARDD | SR-F3 | 61 kDa | NM_032446 | 84466 | 5 | |
| CXCL16 | SR-PSOX | SR-G | 30 kDa | NM_001100812 | 58191 | 17 | miR-451 |
| STAB1 | FEEL-1 | SR-H1 | 276 kDa | NM_015136 | 608560 | 3 | |
| STAB2 | FEEL-2 | SR-H2 | 277 kDa | NM_017564 | 55576 | 12 | |
| CD163 CD163 (soluble) | M130 M130 | SR-I1 SR-I1 | 121 kDa 121 kDa | NM_004244 NM_004244 | 9332 9332 | 12 12 | miR-142-3p miR-181 miR-33a miR-33b |
| RAGE (membrane) | AGER | SR-J1 | 26 kDa | NM_001136 | 177 | 6 | miR-16 miR-214 miR-30 miR221/222 miR-155 |
| RAGE (soluble) | AGER | SR-J1.1 | 14 kDa | AB061668 | 177 | 6 | |
| CD205 | Ly75 | Not known | 198 kDa | NM_002349 | 4065 | 2 | |
| CD206 | MRC1 | Not known | 166 kDa | NM_002438 | 4360 | 10 | miR-124 miR-511-3p |
| CD207 | Langerin | Not known | 37 kDa | NM_015717 | 50489 | 2 | |
| CD209 DC-SIGN | CLEC4L | Not known | 102 kDa | NM_021155 | 30835 | 19 | |
| SRCRB4D | S4D-SRCRB | Not known | 21 kDa | NM_080744 | 136853 | 7 | |
| SSC5D | Not known | Not known | 166 kDa | NM_001144950 | 284297 | 19 | |
| CD14 | Not known | Not known | 11 kDa | NM_000591 | 929 | 5 | miR-21-5p miR-199a-5p miR-199a-3p |
| Current NCBI gene name | ALT names | Proposed nomenclaturea | Protein molecular weight | Accession no. | NCBI gene ID | CHR | ncRNA |
|---|---|---|---|---|---|---|---|
| SCARA1 | SR-A1 CD204 MSR1 | SR-A1 | 50 kDa | NM_138715 | 4481 | 8 | miR-29a miR-155 miR-125b |
| SCARA3 | MSRL1 | SR-A3 | 65 kDa | NM_016240 | 51435 | 8 | |
| COLEC12 | SCARA4 SRCL CL-P1 | SR-A4 | 127 kDa | NM_130386 | 81035 | 18 | |
| SCARA5 | TESR NET33 | SR-A5 | 43 kDa | NM_173833 | 286133 | 8 | |
| MARCO | SCARA2 | SR-A6 | 53 kDa | NM_006770 | 8685 | 2 | |
| SCARB1 | SR-B1 CD36L1 | SR-B1 | 54 kDa | NM_005505 | 949 | 12 | miR-125a miR-455 miR-96 miR-185 miR-223 |
| SCARB2 | LIMP2 CD36L2 LGP85 | SR-B1.1 | 54 kDa | NM_005506 | 950 | 4 | |
| CD36 | SCARB3 FAT GPIV PAS4 | SR-B2 | 88 kDa | NM_001001548 | 948 | 7 | miR-155 miR-27a/b |
| CD68 | gp110 SCARD1 LAMP4 | SR-D1 | 37 kDa | NM_001251 | 968 | 17 | miR-27a miR-146 miR-328 miR-30ea |
| OLR1 | LOX-1 SCARE1 CLEC8A | SR-E1 | 31 kDa | NM_002543 | 4973 | 12 | let-7g miR-29b miR-369-3p miR-370 |
| Dectin 1 | CLEC7A | SR-E2 | 28 kDa | NM_197947 | 64581 | 12 | |
| SCARF1 | SREC-1 | SR-F1 | 87 kDa | NM_003693 | 8578 | 17 | |
| SCARF2 | SREC-II | SR-F2 | 92 kDa | NM_153334 | 91179 | 22 | |
| MEGF10 | EMARDD | SR-F3 | 61 kDa | NM_032446 | 84466 | 5 | |
| CXCL16 | SR-PSOX | SR-G | 30 kDa | NM_001100812 | 58191 | 17 | miR-451 |
| STAB1 | FEEL-1 | SR-H1 | 276 kDa | NM_015136 | 608560 | 3 | |
| STAB2 | FEEL-2 | SR-H2 | 277 kDa | NM_017564 | 55576 | 12 | |
| CD163 CD163 (soluble) | M130 M130 | SR-I1 SR-I1 | 121 kDa 121 kDa | NM_004244 NM_004244 | 9332 9332 | 12 12 | miR-142-3p miR-181 miR-33a miR-33b |
| RAGE (membrane) | AGER | SR-J1 | 26 kDa | NM_001136 | 177 | 6 | miR-16 miR-214 miR-30 miR221/222 miR-155 |
| RAGE (soluble) | AGER | SR-J1.1 | 14 kDa | AB061668 | 177 | 6 | |
| CD205 | Ly75 | Not known | 198 kDa | NM_002349 | 4065 | 2 | |
| CD206 | MRC1 | Not known | 166 kDa | NM_002438 | 4360 | 10 | miR-124 miR-511-3p |
| CD207 | Langerin | Not known | 37 kDa | NM_015717 | 50489 | 2 | |
| CD209 DC-SIGN | CLEC4L | Not known | 102 kDa | NM_021155 | 30835 | 19 | |
| SRCRB4D | S4D-SRCRB | Not known | 21 kDa | NM_080744 | 136853 | 7 | |
| SSC5D | Not known | Not known | 166 kDa | NM_001144950 | 284297 | 19 | |
| CD14 | Not known | Not known | 11 kDa | NM_000591 | 929 | 5 | miR-21-5p miR-199a-5p miR-199a-3p |
ALT, alternative; CHR, chromosome; ncRNA, non-coding RNA.
aSRs that are designated ‘Not known’ were not classified in the regular category and need further study to be named.
Category of SRs in human
| Current NCBI gene name | ALT names | Proposed nomenclaturea | Protein molecular weight | Accession no. | NCBI gene ID | CHR | ncRNA |
|---|---|---|---|---|---|---|---|
| SCARA1 | SR-A1 CD204 MSR1 | SR-A1 | 50 kDa | NM_138715 | 4481 | 8 | miR-29a miR-155 miR-125b |
| SCARA3 | MSRL1 | SR-A3 | 65 kDa | NM_016240 | 51435 | 8 | |
| COLEC12 | SCARA4 SRCL CL-P1 | SR-A4 | 127 kDa | NM_130386 | 81035 | 18 | |
| SCARA5 | TESR NET33 | SR-A5 | 43 kDa | NM_173833 | 286133 | 8 | |
| MARCO | SCARA2 | SR-A6 | 53 kDa | NM_006770 | 8685 | 2 | |
| SCARB1 | SR-B1 CD36L1 | SR-B1 | 54 kDa | NM_005505 | 949 | 12 | miR-125a miR-455 miR-96 miR-185 miR-223 |
| SCARB2 | LIMP2 CD36L2 LGP85 | SR-B1.1 | 54 kDa | NM_005506 | 950 | 4 | |
| CD36 | SCARB3 FAT GPIV PAS4 | SR-B2 | 88 kDa | NM_001001548 | 948 | 7 | miR-155 miR-27a/b |
| CD68 | gp110 SCARD1 LAMP4 | SR-D1 | 37 kDa | NM_001251 | 968 | 17 | miR-27a miR-146 miR-328 miR-30ea |
| OLR1 | LOX-1 SCARE1 CLEC8A | SR-E1 | 31 kDa | NM_002543 | 4973 | 12 | let-7g miR-29b miR-369-3p miR-370 |
| Dectin 1 | CLEC7A | SR-E2 | 28 kDa | NM_197947 | 64581 | 12 | |
| SCARF1 | SREC-1 | SR-F1 | 87 kDa | NM_003693 | 8578 | 17 | |
| SCARF2 | SREC-II | SR-F2 | 92 kDa | NM_153334 | 91179 | 22 | |
| MEGF10 | EMARDD | SR-F3 | 61 kDa | NM_032446 | 84466 | 5 | |
| CXCL16 | SR-PSOX | SR-G | 30 kDa | NM_001100812 | 58191 | 17 | miR-451 |
| STAB1 | FEEL-1 | SR-H1 | 276 kDa | NM_015136 | 608560 | 3 | |
| STAB2 | FEEL-2 | SR-H2 | 277 kDa | NM_017564 | 55576 | 12 | |
| CD163 CD163 (soluble) | M130 M130 | SR-I1 SR-I1 | 121 kDa 121 kDa | NM_004244 NM_004244 | 9332 9332 | 12 12 | miR-142-3p miR-181 miR-33a miR-33b |
| RAGE (membrane) | AGER | SR-J1 | 26 kDa | NM_001136 | 177 | 6 | miR-16 miR-214 miR-30 miR221/222 miR-155 |
| RAGE (soluble) | AGER | SR-J1.1 | 14 kDa | AB061668 | 177 | 6 | |
| CD205 | Ly75 | Not known | 198 kDa | NM_002349 | 4065 | 2 | |
| CD206 | MRC1 | Not known | 166 kDa | NM_002438 | 4360 | 10 | miR-124 miR-511-3p |
| CD207 | Langerin | Not known | 37 kDa | NM_015717 | 50489 | 2 | |
| CD209 DC-SIGN | CLEC4L | Not known | 102 kDa | NM_021155 | 30835 | 19 | |
| SRCRB4D | S4D-SRCRB | Not known | 21 kDa | NM_080744 | 136853 | 7 | |
| SSC5D | Not known | Not known | 166 kDa | NM_001144950 | 284297 | 19 | |
| CD14 | Not known | Not known | 11 kDa | NM_000591 | 929 | 5 | miR-21-5p miR-199a-5p miR-199a-3p |
| Current NCBI gene name | ALT names | Proposed nomenclaturea | Protein molecular weight | Accession no. | NCBI gene ID | CHR | ncRNA |
|---|---|---|---|---|---|---|---|
| SCARA1 | SR-A1 CD204 MSR1 | SR-A1 | 50 kDa | NM_138715 | 4481 | 8 | miR-29a miR-155 miR-125b |
| SCARA3 | MSRL1 | SR-A3 | 65 kDa | NM_016240 | 51435 | 8 | |
| COLEC12 | SCARA4 SRCL CL-P1 | SR-A4 | 127 kDa | NM_130386 | 81035 | 18 | |
| SCARA5 | TESR NET33 | SR-A5 | 43 kDa | NM_173833 | 286133 | 8 | |
| MARCO | SCARA2 | SR-A6 | 53 kDa | NM_006770 | 8685 | 2 | |
| SCARB1 | SR-B1 CD36L1 | SR-B1 | 54 kDa | NM_005505 | 949 | 12 | miR-125a miR-455 miR-96 miR-185 miR-223 |
| SCARB2 | LIMP2 CD36L2 LGP85 | SR-B1.1 | 54 kDa | NM_005506 | 950 | 4 | |
| CD36 | SCARB3 FAT GPIV PAS4 | SR-B2 | 88 kDa | NM_001001548 | 948 | 7 | miR-155 miR-27a/b |
| CD68 | gp110 SCARD1 LAMP4 | SR-D1 | 37 kDa | NM_001251 | 968 | 17 | miR-27a miR-146 miR-328 miR-30ea |
| OLR1 | LOX-1 SCARE1 CLEC8A | SR-E1 | 31 kDa | NM_002543 | 4973 | 12 | let-7g miR-29b miR-369-3p miR-370 |
| Dectin 1 | CLEC7A | SR-E2 | 28 kDa | NM_197947 | 64581 | 12 | |
| SCARF1 | SREC-1 | SR-F1 | 87 kDa | NM_003693 | 8578 | 17 | |
| SCARF2 | SREC-II | SR-F2 | 92 kDa | NM_153334 | 91179 | 22 | |
| MEGF10 | EMARDD | SR-F3 | 61 kDa | NM_032446 | 84466 | 5 | |
| CXCL16 | SR-PSOX | SR-G | 30 kDa | NM_001100812 | 58191 | 17 | miR-451 |
| STAB1 | FEEL-1 | SR-H1 | 276 kDa | NM_015136 | 608560 | 3 | |
| STAB2 | FEEL-2 | SR-H2 | 277 kDa | NM_017564 | 55576 | 12 | |
| CD163 CD163 (soluble) | M130 M130 | SR-I1 SR-I1 | 121 kDa 121 kDa | NM_004244 NM_004244 | 9332 9332 | 12 12 | miR-142-3p miR-181 miR-33a miR-33b |
| RAGE (membrane) | AGER | SR-J1 | 26 kDa | NM_001136 | 177 | 6 | miR-16 miR-214 miR-30 miR221/222 miR-155 |
| RAGE (soluble) | AGER | SR-J1.1 | 14 kDa | AB061668 | 177 | 6 | |
| CD205 | Ly75 | Not known | 198 kDa | NM_002349 | 4065 | 2 | |
| CD206 | MRC1 | Not known | 166 kDa | NM_002438 | 4360 | 10 | miR-124 miR-511-3p |
| CD207 | Langerin | Not known | 37 kDa | NM_015717 | 50489 | 2 | |
| CD209 DC-SIGN | CLEC4L | Not known | 102 kDa | NM_021155 | 30835 | 19 | |
| SRCRB4D | S4D-SRCRB | Not known | 21 kDa | NM_080744 | 136853 | 7 | |
| SSC5D | Not known | Not known | 166 kDa | NM_001144950 | 284297 | 19 | |
| CD14 | Not known | Not known | 11 kDa | NM_000591 | 929 | 5 | miR-21-5p miR-199a-5p miR-199a-3p |
ALT, alternative; CHR, chromosome; ncRNA, non-coding RNA.
aSRs that are designated ‘Not known’ were not classified in the regular category and need further study to be named.
3. Scavenger receptors and microRNAs
3.1 SR-A1
One of the major receptors involved in foam cell formation is macrophage SR class A (SR-A1). It is a trimeric transmembrane glycoprotein consisting of six distinct domains identified first by Kodama et al.7 This receptor mediates the internalization of modified LDL into the cytoplasm of monocytes/macrophages through a specific binding site followed by their transformation into foam cells. These foam cells release growth factors and pro-inflammatory cytokines, and induce an inflammatory milieu.8 While primarily present in monocytes/macrophages, SR-A1 is also present in other cell types, but in small numbers.9 Activation of this receptor has been shown to play an important role in other key processes, such as immune reactions, cell adhesion, and cell–cell interaction, involved in atherogenesis.10–12
The miRs that regulate SR-A1 are mainly focused on its function in relation to inflammation and immune system activation, which are key factors in atherogenesis. A recent study demonstrated that miR-29a increases pro-inflammatory cytokine secretion and SR-A1 expression by targeting the 3′-untranslated region of the lipoprotein lipase (LPL) gene in ox-LDL-stimulated dendritic cells. LPL plays an anti-atherogenic role in the pathogenesis of atherosclerosis as well as in overall lipid metabolism and transport.13,14 Dendritic cells play a central role in the regulation of immunity and influence the initiation and progression of atherosclerosis.15 The inhibition of LPL by ox-LDL-induced overexpression of miR-29a increases SR-A1 and pro-inflammatory cytokine secretion that further stimulates the immuno-inflammatory pathways and atherosclerosis. Interestingly, in this study, the protein expression of other two main SRs, CD36 and lectin-type oxidized LDL receptor 1 (LOX-1), was also increased by miR-29a, while their mRNA profile showed little change. This study showing direct regulation of SR-A1 by miR-29a provides new insights into the role of miR-29a, traditionally thought to be related to fibrosis,16 in the regulation of atherosclerosis. However, it is important to point out that there are many potential targets of miR-29a, and LPL is just one of them and it has a dual role in atherogenesis.14 Thus, further studies on the effect of miR-29a on other factors should be assessed.
In addition to the in vitro study, SR-A1 has also been demonstrated to be closely related to certain miRs in vivo. A group from Tennessee State University found a greater expression of miR-125b in macrophages from SR-A1-null mice than from wild-type (WT) mice subjected to 45 min of myocardial ischaemia followed by reperfusion for up to 7 days.17 The lack of SR-A1 gene and increase of miR-125b resulted in a smaller myocardial infarct size and better cardiac function in the ischaemic mice. To confirm this relationship, they subjected macrophages from SR-A1-null and WT mice to hypoxia followed by reoxygenation and found a significant increase in miR-125b expression in cells from SR-A1-null mice. Importantly, transfection of WT macrophages with miR-125b mimic attenuated cell injury following hypoxia/reoxygenation, and reduced the expression of caspase-3, -7, and -8, p-53, Bak-1, and Bax, all markers of apoptosis. Although the authors did not indicate which gene is the target of miR-125b in this study, their findings suggest a substantial connection between SR-A1 and miR-125b. Since atherosclerosis is the most common cause of myocardial ischaemia, this miR could be potentially used as a biomarker as well therapeutic modality for atherogenesis.
Besides its critical role in atherosclerosis, SR-A1 has also been found to regulate the phagocytic function of alveolar macrophages. Domingo-Gonzalez et al.18 found that syngeneic bone marrow transplant mouse model had an increased SR-A1 profile in the alveolar macrophages. The 3′-untranslated region of SR-AI contains a putative target region for miR-155, and miR-155 expression is decreased post-bone marrow transplant. This observation indicates that regulation of SR-A1 by certain miRs may be the common link between atherosclerosis and other inflammatory disease states.
Regulation of miRs to inhibit SR-A1 may provide a novel strategy to protect against foam cell formation and, thereby, atherogenesis.
3.2 SR-B1
Another type of SRs is SR-B1 that is encoded by the SCARB1 gene. Unlike SR-A1, SR-B1 is a specific receptor for high-density lipoprotein (HDL). SR-B1 facilitates the uptake of cholesteryl esters from HDLs in the liver, which drives the movement of cholesterol from peripheral tissues towards the liver for excretion (reverse cholesterol transport). This ability of SR-B1 makes it a potential target to attenuate or block atherogenesis.19
A number of studies have shown that SR-B1 is regulated by different miRs. Hu et al.20 treated rat adrenals with adrenocorticotropic hormone (ACTH) and primary rat granulosa cells and mouse leydig tumor cells-1 cells with cyclic AMP. ACTH and its second messenger, cyclic AMP, mediate transcriptional regulation of steroidogenic SR-B1 and enhance HDL uptake.21 After treatment with ACTH and cyclic AMP, there was a marked decrease in the expression of miR-125a, -125b, and miR-455, and a reciprocal increase in SR-B1 expression both in rat adrenals and in cultured cells. Using luciferase constructs containing the 3′-untranslated region of SR-B1 combined with miR overexpression and mutagenesis, they provided evidence that steroidogenic SR-B1 is a direct target of miR-125a and miR-455. To further confirm the role of miR-125a and miR-455 in the regulation of SR-B1, they transfected Leydig tumour cells and liver Hepa 1–6 cells with pre-miR-125a and pre-miR-455 (precursor miRs), and found a significant decrease in SR-B1 expression and HDL uptake. These observations suggest that a pivotal role of certain miRs in cholesteryl ester supported steroid hormone production via targeting SR-B1. The deficiency of SR-B1 and HDL may induce atherosclerosis due to their anti-atherogenic role.
Another study by Wang et al.22 showed that in both phorbol myristate acetate-induced human acute monocytic leukemia cell line (THP)-1 cells and ApoE-null mice fed by a high fat diet, SR-B1 and HDL uptake significantly decreased along with an increase in the expression of miR-185, miR-96, and miR-223. Transfection with the mimics of these miRs markedly decreased the expression of SR-B1 and HDL uptake, whereas transfection with inhibitors of these miRs had the opposite effect. The critical effect of miR-185 was further validated by the loss of regulation in constructs with mutated 3′-untranslated region of SR-B1. This study demonstrates that these miRs together directly target SR-BI and alter HDL uptake. Vickers et al.23 demonstrated that SR-B1 was regulated by miR-223 in the ApoE-null mice fed a high fat diet for 16 weeks. The decrease in SR-B1 and increase in miR-223 expression were totally reversed by an intravenous injection of human recombinant HDL (150 μg) collected from patients at the NIH Clinical Center using the cholate dialysis method. Similarly, 3′-untranslated region of SR-B1 was shown to be targeted by miR-223 using a luciferase assay. These results are consistent with another study from the same group, in which both the activity and level of miR-223 promoter were found to be linked to cellular cholesterol homeostasis by targeting SR-B1 in both atheroprone mice and hepatoma cells.24 This effect of miR-223 indirectly contributes to the promotion of ATP-binding cassette transporter A1 expression (mRNA and protein) and enhances cellular cholesterol efflux and decreases HDL expression. Another small study demonstrated that miR-223 has an anti-atherosclerosis effect.25 Importantly, some investigators have shown it to play a key role in inflammatory diseases26 such as atherosclerosis.
These findings suggest that promoting the expression of SR-B1 through modulation of miRs may have a protective effect against atherosclerosis. In essence, SR-B1-related miRs may be of potential therapeutic value in treating atherosclerosis.
3.3 CD36 (SR-B2)
CD36, another member of SR-B family, also known as fatty acid translocase (FAT), has been implicated in cell adhesion, in the phagocytosis of apoptotic cells, and in the metabolism of long-chain fatty acids.27 As a SR, CD36 plays a key role in atherogenesis.28 Ox-LDL is trapped by CD36 on macrophage surface, which, on one hand, provokes signalling cascades that mediate the uptake of ox-LDL and cytokine production. On the other hand, it triggers the activation of mitogen-activated protein kinase family members, such as Lyn, MEKK2, JNK1, and JNK2, and stimulates nuclear factor-kappa B (NF-κB), a transcriptional factor that induces the synthesis of diverse cytokines and regulates the expression of atherosclerosis-related proteins.29
The first study showing that certain miRs might regulate CD36 was a report from Huang et al.30 In their study, exposure of human THP-1 macrophages to ox-LDL led to a marked up-regulation of miR-155 in a dose-dependent manner. Interestingly, silencing of miR-155 with its antisense oligonucleotides significantly increased the expression of both CD36 and CD68. The increased ox-LDL uptake was associated with the release of pro-inflammatory cytokines, such as ILs-6 and -8, and tumour necrosis factor alpha (TNF-α). These effects may be attributed to the enhancement of NF-κB targeted by miR-155, which further induces atherosclerosis. Of note, miR-155 is a protective miR against atherosclerosis. The up-regulation of miR-155 under oxidative stress may be a compensatory mechanism. To confirm this postulate in the in vivo setting, miR-155 was knocked down by locked nucleic acid-anti-miR-155. The blockade of miR-155 resulted in a significant increase in the expression of myeloid differentiation primary response gene 88, an adapter signal used by Toll-like receptors (TLRs) to activate the NF-κB pathway. In another study,31 mice that lacked miR-155 and fed a high fat diet for 6 months showed an increase in CD36 expression, which may due to the reduction in the expression of its target Nr1h3 (liver X receptor alpha, LXRα), a key regulator of macrophage function, which controls the transcriptional programmes with its responsive gene CD36 involved in lipid homeostasis and inflammation.
Recently, Zhang et al.32 reported that miR-27a/b increased the expression of CD36 in THP-1 macrophages treated with ox-LDL by targeting the 3′-UTR site of ATP-binding cassette transporter 1 ABCA1, a major regulator of cellular cholesterol and phospholipid homeostasis. However, this study also showed that miR-27a/b decreases cholesterol efflux to ApoA1 without influencing total cholesterol levels, which may due to the impact of other unknown pathways, and needs further study.
Besides the studies mentioned above, CD36 is also regulated by a variety of miRs under different conditions and in different tissues. Radom-Aizik et al.33 reported that a brief period of exercise may influence 19 miRs in the human blood along with an increase in the expression of CD36, TNF-α, and TLR-4, which may induce rather than reduce atherosclerosis. Another study in human hypertensive kidneys showed that CD36 expression to be decreased in the renal cortex in concert with a change in a number of miRs. However, the authors did not point out the specific miRs which target CD36.34 Naeem et al.35 reported that 14 miRs share in the regulation of a large number of metabolic and immune/oxidative stress target genes, such as CD36, IL-6, and TLR-4, in bovine mammary tissue infected with Streptococcus uberis using bioinformatics analysis.
Taken together, these findings suggest that the inhibitory effect of miRs on CD36 expression may contribute to a reduction in the extent of atherosclerosis. The modulation of these miRs may lead to promising therapies for treating atherosclerotic patients.
3.4 CD68 (SR-D1)
CD68 is another glycoprotein that binds to LDL and is particularly useful as a marker for various cells of macrophage lineage, including monocytes, histiocytes, giant cells, Kupffer cells, and osteoclasts.36 Saha et al.37 reported that miR-27a regulates macrophage differentiation and polarization that plays an important role in inflammatory responses to initiate atherosclerosis;38 transfection of monocytes with anti-miR-27a resulted in reduced CD68 and CD14 expression in alcoholic liver disease though targeting the mRNA of sprouty2, an inhibitor of extracellular signal-regulated kinase (ERK). Since ERK is an important member in inflammatory pathway that participates in atherogenesis, miR-27a based on this finding may have potentially an anti-atherosclerotic function. Butler et al.39 found that seven passenger strand miRs (miR-93*, miR-373*, miR-29b-2*, miR-30c-1*, miR-27a*, miR-27b*, and miR-149*) were disturbed in association with six separate down-regulated target genes (CD68, FAM102A, MXI1, MYO1D, TP53INP1, and ZRANB1) including CD68 in obesity-related syndromes such as Prader–Willi syndrome and Alström syndrome. Several investigators have reported that CD68 may be related to different miRs in macrophages in other diseases, such as RA (miR-146) and gastrointestinal cancer (miR-30e* and miR-328).40–42 Although these studies39–42 did not find a direct link between CD68 and miRs in relation to atherosclerosis, these miRs might influence inflammation and oxidative stress, which are significant contributors in atherogenesis.
Collectively, these studies suggest that CD68 is directly or indirectly regulated by miRs. These miRs may have potential anti-atherosclerosis activity by repressing CD68 expression.
3.5 LOX-1 (SR-E1)
LOX-1, identified by Sawamura et al.43 with further work on its biology by our group,44 belongs to the C-type lectin superfamily and plays a major role as a SR through binding to ox-LDL. It is expressed on the membrane of different cell types, including endothelial cells, vascular smooth muscle cells (VSMCs), fibroblasts, and macrophages. Its expression is stimulated by TNF-α, angiotensin II, shear stress, and ox-LDL itself. The up-regulation of LOX-1 subsequently triggers reactive oxygen species (ROS) generation through activation of NADPH oxidases, followed by stimulation of redox signals, such as MAPK p38, ERK1/2, and JNK, which promote the activation of transcriptional factor NF-κB that navigates the expression of proteins with a major role in atherogenesis.44
Recent studies from our laboratory have shown that LOX-1 expression in response to ox-LDL is inhibited by let-7g in a time- and dose-dependent fashion in human aortic SMCs.45 Furthermore, LOX-1-null mice fed a high cholesterol diet were shown to have increased let-7g expression.46 These results are consistent with a study by Chen et al.47 who showed a let-7g-binding site on the 3′-untranslated region of LOX-1 mRNA in human SMCs. In addition to let-7g, LOX-1 has been found to be regulated by miR-29b in the aorta.48 In this study, miR-29b expression was increased by ox-LDL in the aorta of hyperlipidaemic mice, along with enhancement of ROS generation and expression of its downstream signalling MAPKs (ERK1/2). These effects of miR-29b were attenuated by LOX-1 knockdown (using LOX-1 shRNA). Using the promoter reporter assay and chromatin immunoprecipitation, they found that activator protein-1, a regulator of LOX-1, is the target of miR-29b. Of note, only miR-29b-1/miR-29a cluster gene found on chromosome 7 was found to regulate LOX-1, but not the other distinct miR-29b gene located on chromosome 1. These results taken together suggest that ox-LDL enhances miR-29b expression via the LOX-1/ROS/ERK signalling pathway. These data are similar to those reported by Chen et al.,13 who observed LOX-1 to be increased by miR-29a.
LOX-1 is also regulated by miRs in other disease states in different species. Serpente et al.49 demonstrated an association between +1073 C/T, a rs1050283 single-nucleotide polymorphism located in the 3′-untranslated region of LOX-1 and decreased LOX-1 protein levels in peripheral mononuclear blood cells from 20 Alzheimer's disease (AD) patients; these authors also showed regulation of LOX-1 by has-miR-369-3p. This association was confirmed in a meta-analysis in which LOX-1 was found to be affected by many miRs including has-miR-369-3p.50 Another interesting study performed in Bos taurus demonstrated that LOX-1 is associated with milk production and health traits in dairy cattle through binding to bta-miR-370.51
These findings indicate that LOX-1 is modulated by several different miRs in a variety of diseases in different species. However, none of these studies were performed in macrophages which are key cell line in atherogenesis. Furthermore, there is no information on factors that induce the change/s in these miRs. Thus, future studies should focus on the role of miRs on LOX-1 expression in models of atherosclerosis.
3.6 CXCL16 (SR-G) and CD14
CXC chemokine ligand 16 (CXCL16) is a CXC soluble chemokine characterized as an adhesion molecule and belonging to the SRG family that regulates inflammation, tissue injury, and fibrosis.52 A study shows that elevation of serum CXCL16 level correlates with acute ischaemic stroke in patient with atherosclerosis of the carotid artery.53 There is no direct evidence as yet showing miRs controlling CXCL16 expression in atherosclerosis, except that a clinical study reported CXCL16 to be down-regulated by miR-451 in patients with osteosarcoma. Furthermore, an in vitro study showed that silencing of CXCL16 could phenocopy the effects of miR-451 on phenotypes of osteosarcoma cells.54 Although there is no direct evidence showing that CXCL16 is regulated by miRs in atherosclerosis, its character of SR still makes it as a potential target for miR-based biomarker in atherogenesis.
CD14 is mainly expressed on macrophages. It acts as a co-receptor (along with the TLR4) for the detection of bacterial lipopolysaccharide. It can bind lipopolysaccharide only in the presence of lipopolysaccharide-binding protein. Activation of TLR4 and CD14 leads to downstream release of inflammatory modulators, including TNF-α and IL-1, which subsequently trigger inflammatory signals such as NF-κB and MAPKs. Recently, CD14 has been described to be a potential biomarker for the diagnosis of stable coronary artery disease.55 Using a CD14 knockdown RAW264.7 macrophage cell line, Du et al.56 found that lack of CD14 significantly changed the expression of miR-199a-3p, miR-199a-5p, and miR-21-5p when cells were treated with lipopolysaccharide. However, the authors did not define the target of these miRs, and the underlying mechanism thus remains unclear.
Collectively, both CXCL16 and CD14 appear to be regulated by miRs. However, the evidence of their anti-atherosclerosis function needs further proof.
3.7 RAGE (SR-J1)
Receptor for advanced glycation end-products (RAGEs), also known as SR-J1, is a member of SR superfamily, and represents the metabolic memory underlying the pathophysiology of chronic diabetic complications. AGEs trigger intracellular damage through stimulating RAGE signalling that leads to the elevation of cytosolic ROS, activation of NF-κB, and induction of oxidative and endoplasmic reticulum stress that are key processes in atherogenesis.57 Recent studies have identified a series of miRs involved in the regulation of AGE/RAGE signalling in micro- and macrovascular complications of diabetes.58 Shanmugam et al.59 found that in THP-1 monocytes, the RAGE ligand, S100b, significantly down-regulated the expression of miR-16 that may destabilize COX-2 mRNA by binding to its 3′-untranslated region. The up-regulation of the pro-inflammatory COX-2 gene may induce inflammation and atherosclerosis in diabetics. The inhibition or enhancement of miR-16 expression with its inhibitor or mimic oligonucleotides was shown to significantly increase or decrease COX-2 mRNA expression, respectively, which indicates that COX-2 may be a direct target of miR-16. This study suggests that key RNA-binding proteins and miRs may be used to prevent the effect of RAGEs on COX-2 to efficiently stabilize inflammatory genes via opposing the actions of key RNA-binding proteins and miRs.
The basis of AGEs inducing inflammation is thought to be their ability to delaying spontaneous apoptosis of monocytes. However, the underlying mechanism was not clear until a recent study showed that the phosphatase and tensin homologue (PTEN), an apoptosis-related gene, is inhibited by miR-214 that is up-regulated by AGEs via RAGE in THP-1 cells.60 Hilgendorf et al.61 have shown that the increased secretion of miR-214 by AGEs inhibits PTEN expression and induces monocyte survival, which drives progression of atherosclerosis.
In addition to miR-16 and miR-214, other miRs, such as miR-30 family and miR-221/222, have also been shown to be related to RAGE in renal disease and cancers. Shi et al.62 observed in mutant mice with proteinuria and glomerulosclerosis that RAGE is up-regulated by the inhibition of miR-30 family, including miR-30b, miR-30c-1, miR-30c-2, and miR-30d. Furthermore, RAGE has been reported to be involved in human papillary thyroid cancer, in which miR-221/222 is up-regulated by high-mobility group box-1 (HMGB1) protein, a late inflammatory cytokine related to RAGE.63 Interestingly, miR-221/222 has also been found to target nitric oxide (NO) synthase which has two opposite effects in the atherosclerotic process,64 which may indicate a complicated role of miR-221/222 in the expression of RAGE and the development of atherosclerosis. Another study suggested that RAGE up-regulates miR-155 expression via AP-1 activation in colon cancer, since the blockade of RAGE with anti-RAGE antibody suppressed the induction of miR-155 by exogenous S100P, a stimulus of RAGE.65
These observations collectively suggest that RAGE by modulation of specific miRs could be a target for therapy of atherosclerosis and related disorders. Although some miRs are not found to be related to RAGE in atherosclerosis, understanding the role of RAGE and related miRs may enhance our knowledge of atherogenesis.
3.8 CD163 (SR-I1) and CD206
CD163 and CD206 are highly expressed on haemoglobin-associated macrophages [M(Hb)] that are distinct from traditional macrophage foam cell forming macrophages due to the lack of lipid deposits or production of pro-inflammatory cytokines. Unlike other inflammatory SRs, CD163 and CD206 produce anti-inflammatory cytokines, such as IL-10, that play an inhibitory role in atherosclerosis.66
Growing body of evidence shows that CD163 and CD206 are regulated by miRs. Lagrange et al.67 found a dramatic decrease in the expression of haematopoietic-specific miR-142-3p, along with a decrease in CD163 and CD16 expression, during differentiation of human monocytes into macrophages induced by colony-stimulating factor 1 (CSF-1). Up- and down-regulation of miR-142-3p significantly changed CD163 expression through encoding the transcription factor—early growth response 2 (EGR2), a direct target of miR-142-3p. Recently, Gao et al.68 showed that miR-181 can inhibit the replication of porcine reproductive and respiratory syndrome (PRRSV) virus, through inhibiting its genomic RNA. In this study, as a receptor of PRRSV, CD163 was found to be bound to miR-181 with 3′-untranslated region of its mRNA in blood monocytes and porcine alveolar macrophages. Since the monocytes and macrophages play a key role in atherogenesis, future studies are necessary to show that these miRs regulate CD163.
In addition to the two miRs mentioned here, CD163 has been shown to be closely related to miR-328 down-regulation in tumour-infiltrating macrophages from patients with surgically resected gastric cancer.42 These studies demonstrated that CD163 is directly or indirectly regulated by a variety of miRs. Interestingly, the soluble CD163, a marker of macrophage polarization, is elevated rather than depressed in atherosclerosis, and has been shown to be related to an increase in miR-33a (≈4 times) and miR-33b (≈3 times), respectively, in obese children.69,70 These findings strongly suggest that CD163 is involved in the process of atherogenesis via specific miRs.
Another anti-atherosclerosis SR CD206 is also regulated by miRs. In studies on inflammation and tumours, Veremeyko et al.71 demonstrated that exposure of macrophages to IL-4 and IL-13 to induce their polarization towards the M2 phenotype significantly increased miR-124 expression. Up-regulation of M2 marker, such as CD206, was abrogated by an miR-124 inhibitor, suggesting that miR-124 contributed to the M2 phenotype development and to the maintenance of CD206. Squadrito et al.72 showed that miR-511-3p is encoded by both the mouse and human CD206 gene, and is dramatically increased with the overexpression of CD206 in tumour-associated macrophages. However, enforcing miR-511-3p expression did not increase the expression of CD206 or the tumour growth, which indicates a complicated relationship between miR-511-3p and CD206. Since M2 polarization negatively controls atherogenesis, the up-regulation of its marker CD206 by miR-124 and miR-511-3p may have potential anti-atherosclerotic effects.
The relationships between different SRs and miRs in atherogenesis are summarized in Figure 1. As discussed earlier, inhibition of SRs is an important method to inhibit atherosclerosis. Utilizing an miR-based strategy is an efficient way to regulate SRs. However, current knowledge of the biology of different SRs is still far from complete. For example, we have recently observed that the up-regulation of some SRs may induce down-regulation of the other SRs.73 Thus, the change of a single miR may not be able to totally overcome the impact of other SRs in atherosclerosis. Obviously, this area needs further investigations to explore the potential therapeutic value of miRs.
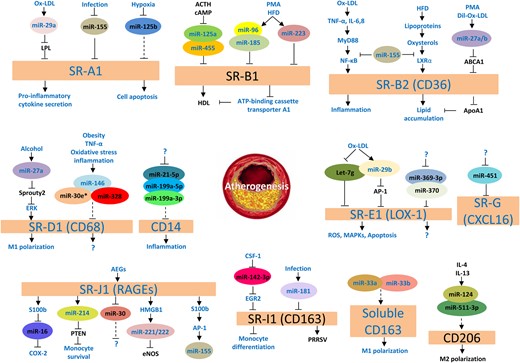
Relationship between SRs and miRs. Names in oval boxes refer to miRss; arrows depicting activation or inhibition between the indicators mean that the upper indicator normally activates or inhibits the one below; blue font or black font colour indicates that this indicator is up-regulated or down-regulated in atherosclerosis; solid line implies that the relationship between two indicators is direct, and dotted line implies that the relationship between two indicators is indirect in the development of atherosclerosis; question mark refers to unknown indicator. Ox-LDL, oxidized low-density lipoprotein; LPL, lipoprotein lipase; SRs, scavenger receptors; ACTH, adrenocorticotropic hormone; cAMP, cyclic adenosine monophosphate; HDL, high-density lipoprotein; PMA, phorbol myristate acetate; HFD, high fat diet; TNF-α, tumour necrosis factor alpha; IL, interleukin; LXRα, liver X receptor alpha; ABCA1, ATP-binding cassette transporter-1; APOA1, apolipoprotein A-1; ERK, extracellular signal-regulated kinase; M1, macrophage-1; AP-1, activator protein-1; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1; ROS, reactive oxygen species; MAPKs, mitogen-activated protein kinases; AEGs, advanced glycation end-products; RAGEs, receptor for advanced glycation end-products; COX-2, cyclooxygenase-2; PTEN, phosphatase and tensin homologue; HMGB1, high-mobility group box-1; eNOS, endothelial nitric oxide synthase; CSF-1, colony-stimulating factor 1; EGR2, early growth response 2; PRRSV, porcine reproductive and respiratory syndrome; M2, macrophage 2.
3.9 Scavenger receptors and long ncRNAs
Another large group of ncRNAs are the so-called lncRNAs that consist of >200 nucleotides. The lncRNAs regulating genes can be categorized into six groups: (i) sense: overlapping and sharing the same promoter as a protein-coding gene; (ii) antisense: located in antisense orientation to a protein-coding gene. (iii) Intronic: located in an intron of a protein-coding gene. (iv) Intergenic: located in between two protein-coding genes. (v) Enhancer: located in the enhancer region of a protein-coding gene. (vi) Circular: forms a covalently enclosed circular RNA that arises from splicing of a protein-coding gene.74
To date, direct evidence of lncRNA regulating SRs has not been presented. However, a number of studies suggest that lncRNAs are involved in the inflammatory cascade and atherosclerosis. As SR activation plays a key role in the inflammation, and subsequently atherogenesis, it is reasonable to hypothesize that SRs may participate in lncRNA-related atherosclerosis, and may link with yet undefined lncRNAs. In the subsequent paragraphs, we review relatively recent studies that link lncRNAs in inflammation and atherosclerosis.
The first lncRNA found in relation to atherosclerosis was ANRIL, a neighbour of tumour suppressor CDKN2A/B encoded in the human chromosome 9p21 region.75 Congrains et al.76 found that the increased expression of ANRIL was consistently associated with the risk alleles for atherosclerosis-related phenotypes and common carotid artery stenosis. Abrogation of ANRIL with its siRNA caused a slightly but significant reduction of CDKN2A and an induction of CDKN2B and VSMC growth, which promotes atherosclerosis. The function of ANRIL in atherosclerosis is mainly based on its exons1–2 site.77 Other evidence was presented by Holdt et al.78 who demonstrated that ANRIL regulates target genes in trans, leading to increased SMC proliferation, enhanced cell adhesion, and decreased apoptosis, which are all essential features of atherogenesis. This effect of transregulation was dependent on Alu motifs, markers for the promoters of ANRIL target genes, which provides a molecular mechanistic basis for the proatherogenic effects of ANRIL. Moreover, ANRIL appears to be negatively correlated with ADIPOR1, VAMP3, and C11ORF10, genes with a well-described role in fatty acid oxidation and glucose metabolism, as well as in inflammation. This again indicates that ANRIL may have a close relationship with atherosclerosis.79
Recently, a study by Wu et al.80 identified lincRNA-p21 as a key regulator of cell proliferation and apoptosis during atherosclerosis. In their study, atherosclerotic plaques in ApoE-null mice showed a significantly decreased expression of lincRNA-p21. In their concurrent in vitro study, inhibition of lincRNA-p21 markedly induced proliferation and repressed apoptosis of VSMCs and macrophages. Patients with coronary artery disease showed decreased expression of lincRNA-p21. Moreover, the genome-wide analysis revealed that p53, a regulator of apoptosis, decreased along with the down-regulation of lincRNA-p21. Since the inactivation of p53 stimulates the development of atherosclerosis through promotion of the expression of mouse double minute 2, an E3 ubiquitin-protein ligase, the down-regulation of lincRNA-p21 may be associated with enhanced atherosclerosis.
Another study performed in human acute monocytic leukaemia macrophage-derived foam cells showed an increase in lncRNA RP5-833A20.1, and a decrease in nuclear factor IA (NFIA) expression.81 The decreased NFIA expression may be related to the up-regulation of hsa-miR-382-5p expression, which is downstream of RP5-833A20.1. Importantly, the RP5-833A20.1/hsa-miR-382-5p/NFIA pathway influenced the regulation of cholesterol homeostasis (decreased HDL-C, increased LDL-C, and VLDL-C) and inflammatory responses (increased expression of IL-1β, IL-6, TNF-α, and C-reactive protein) in ApoE-null mice. Importantly, they also predicted that SRs, such as SR-A1 and CD36, may be regulated by this pathway, although no direct evidence was given. In another study, this group identified another lncRNA, DYNLRB2-2, to be significantly induced by ox-LDL, which promoted ABCA1-mediated cholesterol efflux and inhibited inflammation through G protein-coupled receptor 119 (GPR119), a regulator of glucose and lipid metabolism, in THP-1 macrophage-derived foam cells. Of note, the enhancement of GPR119 decreased lipid and serum inflammatory cytokine levels and prohibited atherosclerosis in ApoE-null mice, probably by a negative feedback mechanism.82
Endothelial dysfunction is another important cause for atherosclerosis, which is characterized as an imbalance of endothelial NO synthase (NOS) activity and NO formation/release. Studies from Marsden laboratory showed that lncRNA NAT sONE, an eNOS antisense, is induced by hypoxia in endothelial cells and it subsequently reduces eNOS expression via overlapping its mRNA transcripts, which indicates an lncRNA-inducer effect in the formation of atherosclerosis.83,84 Another recent study showed that lncRNA, an apolipoprotein A-I (APOA1) antisense, inhibits the expression of APOA1, a major protein component of HDL, in both in vitro and in vivo studies.85 Since HDL is a protective lipoprotein for atherosclerosis, its inhibition by APOA1 antisense may promote atherogenesis.
Taken together, early work discussed here suggests that lncRNAs may be important players in atherosclerosis. These lncRNAs can affect several key processes in atherogenesis, including VSMC proliferation, endothelial dysfunction, lipid metabolism, and inflammation. LncRNAs involved in atherosclerosis are summarized in Supplementary material online, Figure S1.
4. Conclusion and future perspectives
This review summarizes the information on ncRNAs and SRs directly or indirectly related to atherogenesis. As shown in Figure 1, Table 2, and Supplementary material online, Figure S1, some traditional factors in atherogenesis, such as oxidative stress, inflammation, hypoxia, and high cholesterol intake, are responsible for changes in miRs and lncRNAs. There may well be other factors in this process. Changes in the profile of SRs and ncRNAs in atherosclerosis are based on several pathways, including cell apoptosis, macrophage polarization, monocyte differentiation, endothelial dysfunction, VSMC proliferation, MAPKs, lipid homeostasis, and inflammation pathways. Since these pathways are also involved in other diseases such as AD, hepatosteatosis, pulmonary disease, and many cancers, relationship between SRs and ncRNAs described in this review suggests that they may act as a bridge between atherosclerosis and other disease states, especially cancers.
LncRNAs in atherosclerosis
| LncRNA | Correlation | Function | Role in AS | References |
|---|---|---|---|---|
| ANRIL | CDKN2A/B | Increases VSMC growth | Syn | 76,77 |
| Alu motifs | Promotes cell proliferation, cell adhesion, and apoptosis | Syn | 78 | |
| ADIPOR1, VAMP3, C11ORF10 | Promotes fatty acid oxidation, glucose metabolism, and inflammation | Syn | 79 | |
| lincRNA-p21 | p53 | Decreases cell proliferation and induces apoptosis | Anta | 80 |
| RP5-833A20.1 | NFIA | Decreases HDL-C, increases LDL-C and VLDL-C, IL-1β, IL-6, TNF-α, and CR-P | Syn | 81 |
| DYNLRB2-2 | GPR119 | Inhibits lipid and serum inflammatory cytokine levels | Anta | 82 |
| NAT sONE | eNOS | Decreases eNOS expression and induces EC dysfunction | Syn | 83,84 |
| APOA1 antisense | APOA1 | Decreases HDL-C | Syn | 85 |
| LncRNA | Correlation | Function | Role in AS | References |
|---|---|---|---|---|
| ANRIL | CDKN2A/B | Increases VSMC growth | Syn | 76,77 |
| Alu motifs | Promotes cell proliferation, cell adhesion, and apoptosis | Syn | 78 | |
| ADIPOR1, VAMP3, C11ORF10 | Promotes fatty acid oxidation, glucose metabolism, and inflammation | Syn | 79 | |
| lincRNA-p21 | p53 | Decreases cell proliferation and induces apoptosis | Anta | 80 |
| RP5-833A20.1 | NFIA | Decreases HDL-C, increases LDL-C and VLDL-C, IL-1β, IL-6, TNF-α, and CR-P | Syn | 81 |
| DYNLRB2-2 | GPR119 | Inhibits lipid and serum inflammatory cytokine levels | Anta | 82 |
| NAT sONE | eNOS | Decreases eNOS expression and induces EC dysfunction | Syn | 83,84 |
| APOA1 antisense | APOA1 | Decreases HDL-C | Syn | 85 |
Syn means that this lncRNA regulates atherogenesis in a synergistic manner; Anta means that this lncRNA regulates atherogenesis in an antagonistic manner.
lncRNA, long non-coding RNAs; ANRIL, antisense non-coding RNA in the INK4 locus; VSMCs, vascular smooth muscle cells; ADIPOR1, Adiponectin receptor 1; VAMP3, vesicle-associated membrane protein 3; NFIA, nuclear factor IA; HDL-C, high-density lipoprotein-C; LDL-C, low-density lipoprotein-C; VLDL-C, very low-density lipoprotein-C; IL-1β, interleukin-1 beta; IL-6, interleukin-6; TNF-α, tumour necrosis factor alpha; CR-P, C-reactive protein; GPR119, G protein-coupled receptor 119; eNOS, endothelial nitric oxide synthase; EC, endothelial cell; APOA1, apolipoprotein A-I.
LncRNAs in atherosclerosis
| LncRNA | Correlation | Function | Role in AS | References |
|---|---|---|---|---|
| ANRIL | CDKN2A/B | Increases VSMC growth | Syn | 76,77 |
| Alu motifs | Promotes cell proliferation, cell adhesion, and apoptosis | Syn | 78 | |
| ADIPOR1, VAMP3, C11ORF10 | Promotes fatty acid oxidation, glucose metabolism, and inflammation | Syn | 79 | |
| lincRNA-p21 | p53 | Decreases cell proliferation and induces apoptosis | Anta | 80 |
| RP5-833A20.1 | NFIA | Decreases HDL-C, increases LDL-C and VLDL-C, IL-1β, IL-6, TNF-α, and CR-P | Syn | 81 |
| DYNLRB2-2 | GPR119 | Inhibits lipid and serum inflammatory cytokine levels | Anta | 82 |
| NAT sONE | eNOS | Decreases eNOS expression and induces EC dysfunction | Syn | 83,84 |
| APOA1 antisense | APOA1 | Decreases HDL-C | Syn | 85 |
| LncRNA | Correlation | Function | Role in AS | References |
|---|---|---|---|---|
| ANRIL | CDKN2A/B | Increases VSMC growth | Syn | 76,77 |
| Alu motifs | Promotes cell proliferation, cell adhesion, and apoptosis | Syn | 78 | |
| ADIPOR1, VAMP3, C11ORF10 | Promotes fatty acid oxidation, glucose metabolism, and inflammation | Syn | 79 | |
| lincRNA-p21 | p53 | Decreases cell proliferation and induces apoptosis | Anta | 80 |
| RP5-833A20.1 | NFIA | Decreases HDL-C, increases LDL-C and VLDL-C, IL-1β, IL-6, TNF-α, and CR-P | Syn | 81 |
| DYNLRB2-2 | GPR119 | Inhibits lipid and serum inflammatory cytokine levels | Anta | 82 |
| NAT sONE | eNOS | Decreases eNOS expression and induces EC dysfunction | Syn | 83,84 |
| APOA1 antisense | APOA1 | Decreases HDL-C | Syn | 85 |
Syn means that this lncRNA regulates atherogenesis in a synergistic manner; Anta means that this lncRNA regulates atherogenesis in an antagonistic manner.
lncRNA, long non-coding RNAs; ANRIL, antisense non-coding RNA in the INK4 locus; VSMCs, vascular smooth muscle cells; ADIPOR1, Adiponectin receptor 1; VAMP3, vesicle-associated membrane protein 3; NFIA, nuclear factor IA; HDL-C, high-density lipoprotein-C; LDL-C, low-density lipoprotein-C; VLDL-C, very low-density lipoprotein-C; IL-1β, interleukin-1 beta; IL-6, interleukin-6; TNF-α, tumour necrosis factor alpha; CR-P, C-reactive protein; GPR119, G protein-coupled receptor 119; eNOS, endothelial nitric oxide synthase; EC, endothelial cell; APOA1, apolipoprotein A-I.
SRs play key roles in inflammatory diseases like atherosclerosis. The new knowledge of epigenetics provides an opportunity to regulate atherogenesis and its consequences by diverse array of ncRNAs. However, some SRs provided in Table 1, such as COLEC12, SCARA5, MARCO, Dectin 1, etc., have not been shown to be regulated by ncRNAs. Furthermore, most studies in this article have focused on the effect of SRs and ncRNA on certain disease states. The direct impact of disease states, such as angioplasty or stenting of coronary arteries, or treatment on SRs and ncRNA is not known. Finally, our current knowledge of lncRNAs and miRs, and their relationship with SRs, is still in its infancy. There is still a long way to go one ncRNA-based therapy due to a series of limitations: (i) the biology of ncRNAs is still not fully elucidated; (ii) a single miR could regulate several SRs and vice versa. This implies that the specific miR therapy may not completely block SR and may have unknown side-effects; (iii) there are interactions between different miRs, and miRs and lncRNAs, which make the ncRNA-based therapeutic approach more complex; (iv) the poor conservation and the fact that most lncRNAs are expressed as various transcript variants challenge the identification of specific biological functions on SRs; (v) almost all existing miRNA target prediction methods neglected the influence of the cellular environment. This deficiency may be overcome by a new method called cell type-specific miR occupancy rate (MIROR) prediction.86 Therefore, much work needs to be done to determine the precise functions of ncRNAs, particularly with reference to regulation of SRs and the development of atherosclerosis, before using them as biomarkers or as therapeutic modality.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This study was supported by funds from the National Natural Science Foundation of China (grant no. 81500344); Anhui Provincial Natural Science Foundation (grant no.1508085MH178); and the National Natural Science Foundation for Fostering Young Scholars of China (the First Affiliated Hospital of Anhui Medical University, grant no. 2013KJ25). Additional funding was provided by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, Washington, DC (Merit Review grant to J.L.M.).
References

{kind=link}