





Abstract
We determined the contribution of the desmosomal cadherin desmoglein-2 to cell–cell cohesion in cardiomyocytes. In the intercalated disc, providing mechanical strength and electrical communication between adjacent cardiomyocytes, desmoglein-2 is closely associated with N-cadherin and gap junctions.
We studied intercalated discs of HL-1 cardiomyocytes by immunostaining of desmoglein-2 and N-cadherin. Cohesion was measured using a liberase-based dissociation-assay and compared with cell-free single-molecule atomic force microscopy measurements. l-tryptophan caused irregular desmoglein-2 condensation, weakened cell–cell cohesion and impaired both homophilic desmoglein-2 and N-cadherin trans-interaction, whereas l-phenylalanine had no effect. l-tryptophan did not affect N-cadherin localization and its inhibitory effect on cell-cohesion and desmoglein-2 binding, but not on N-cadherin interaction, was blocked by a desmoglein-specific tandem peptide. Moreover, Ca2+-depletion, desmoglein-2 knockdown, a desmoglein-specific single peptide and certain desmoglein-2 mutations associated with arrhythmogenic cardiomyopathy reduced cell–cell cohesion, whereas cell adhesion was strengthened by desmoglein-2 overexpression. Since single peptide did not interfere with N-cadherin trans-interaction, these data indicate that (i) desmoglein-2 binding is crucial for cardiomyocyte cohesion and (ii) l-tryptophan reduced both desmoglein-2 and N-cadherin binding, whereas single and tandem peptide can be used to specifically target desmoglein-2-mediated adhesion. l-tryptophan and single peptide also induced ultrastructural alterations of areae compositae. Functional analyses at the organ level revealed reduced cardiomyocyte function and inefficient response to adrenergic stimulation in both l-tryptophan- and single peptide-challenged murine Langendorff hearts paralleled by redistribution of connexin 43 in l-tryptophan-treated heart slices.
Our data demonstrate that desmoglein-2 plays a critical role in cardiomyocyte cohesion and function.
1. Introduction
Arrhythmogenic cardiomyopathy (AC) is a hereditary heart muscle disease caused by mutations of genes encoding components of the desmosome.1 The prevalence of AC ranges between 1:1000 and 1:5000 with huge regional variance.2 Although AC is a relatively rare disease its impact on society is considerable, because it affects patients early in life and commonly leads to sudden cardiac death without previous symptoms.3 Mutations in 12 different genes have been linked with AC development, among which are genes encoding for the desmosomal proteins plakoglobin (PG), desmoplakin (Dsp), plakophillin-2 (Pkp2), desmoglein-2 (Dsg2), and desmocollin-2 (Dsc2).4
In the heart, unlike in other desmosome-containing tissues like the skin, desmosomes form a close association with adherens junctions and neighbouring gap junctions. Although some areae compositae look more desmosome-like and others more adherens junction-like, both types of junction possess Dsg2.5 This special structure the area composita, or intercalated disc (ICD), resides at the short end of cardiomyocytes and links neighbouring cells mechanically as well as electrically. This cell–cell contact consists of an extracellular part, in which N-cadherin (N-Cad), Dsg2, and Dsc2, as well as connexin 43 are linked with their counterparts in a trans-interaction across the cell membranes of adjacent cardiomyocytes. On the cytosolic side, linker proteins such as PG, Dsp, and Pkp2 connect the areae compositae to the actin cytoskeleton and the desmin intermediate filament system to provide mechanical strength. Recently, it has become obvious that the area composita not only fulfils mechanical functions via its adherens junction- and desmosome-like parts and electrical functions via its gap junctions, but also plays a role in cell–cell communication via influencing adjacent ion channels and signalling pathways.6,7
Dsg2 is the only desmoglein expressed in the heart and performs heterodimerization with Dsc2 to provide cell–cell stability. Therefore, in the heart there is no possibility for the compensation of Dsg2 loss-of-function by other desmoglein subtypes. Dsg2 mutations have been demonstrated in 10% of AC patients and more than 50 pathogenic mutations of the gene are known.4
Currently, mutations associated with AC are studied by sequencing approaches in AC families and in some studies the mutated protein was expressed in vitro to research its interaction with other area composita members, e.g. in yeast-two-hybrid or co-immunoprecipitation assays.8 More complex and time-consuming approaches involve expression of the mutated desmosomal component in animal models,9,10 or non-cardiac cell lines.11 However, effective in vitro tests for functional changes in cardiomyocytes expressing these mutants are necessary.
Here, we demonstrate that inhibition of desmoglein interaction as well as specific targeting of Dsg2 in vitro results in structural and functional changes in cardiomyocytes. We demonstrated condensation of Dsg2, but not N-Cad, after inhibition of extracellular interaction of desmoglein. A functional HL1-cardiomyocyte cohesion assay and ultrastructural analyses revealed reduced cell–cell contact strength upon inhibition of Dsg2 binding. We furthermore showed that the Dsg2 mutations A517V and V920G led to a loss of cardiomyocyte-cohesion. Targeting of cadherin–cadherin interaction on the organ level induced an ICD tear with lateralization of connexin 43 and disruption of beta1-adrenergic receptor localization which may account for a reduction of heart rate, cardiac performance, and response to adrenergic challenging in the isolated mouse heart.
2. Methods
2.1 Cell culture
HL-1 cells, a murine atrial cardiomyocyte cell line, were kindly provided by William C. Claycomb (New Orleans, USA) and cultured according to his instructions. Further details on cultivation and expression of exogenous DNA or siRNA as well as reagents used for cell treatment are provided in Supplementary material online, Methods.
2.2 Immunostaining
Immunostaining of HL-1 cells was performed as previously described.12 Details on antibodies used are provided in Supplementary material online, Methods. Particle size was determined using the Analyze Particles function of Image J software and the mean size of areas with Dsg2-positive adjacent pixels was measured and compared with PBS as a control, which was normalized to 100%.
2.3 Western blot analysis
Western blot analysis was performed using standard procedures, details are provided in Supplementary material online, Methods.
2.4 Liberase assay
HL-1 cells were grown in coated 24-well-plates for 7 days and transfected with Dsg2 mutants/siRNA or treated with antibodies/chemicals for time-periods indicated. Cell-monolayers were washed with PBS and incubated with 150 µL liberase buffer (liberase DH 1.3 units/mL, Roche, Mannheim, Germany; dispase II 2.5 units/mL, Sigma-Aldrich, St Louis, USA, in Hanks’ balanced saline solution, HBSS, AppliChem with 4.2 mM NaHCO3 and 0.13 mM CaCl2 added) per well at 37°C for 30 min to detach the HL-1 cell monolayer from the well bottom. After detachment, 200 µL of HBSS was added to each well and the cell-monolayers were subjected to shear stress by pipetting three times using an automated pipette. The total number of resulting fragments per well was determined using a binocular stereo microscope (Leica). Fragments were counted if they were clearly visible at 30-fold magnification. In Supplementary material online, Figure S1, we show a representative area in a part of each well. Strong fragmentation after shear stress is an indicator of weakened cell–cell cohesion. This assay facilitates functional measurements of cell cohesion in HL-1 cardiomyocytes. The assay is reliable and simple to perform and has been used previously by us and others to measure keratinocyte cell–cell adhesion.13–16 A limitation of the method is that it measures overall cell cohesion, which by itself does not allow conclusions on the contribution of a specific adhesion molecule. To overcome this limitation, the assay should be evaluated in parallel with more specific molecular approaches such as single molecule atomic force measurements as in this study. Caution should be taken on enzyme concentrations as activities can vary with charge.
2.5 Atomic force microscopy measurements of Dsg2 and N-Cad interactions
We applied a Nanowizard III AFM (JPK Instruments, Berlin, Germany) to study the effects of SP, TP, and L-Tryp on Dsg2 and N-Cad trans-adhesion in vitro. Purification, sample preparation, and measurements are detailed in Supplementary material online, Methods and were performed essentially as described elsewhere.17
2.6 Electron microscopy
Confluent HL-1 cell monolayers were fixed with glutaraldehyde, embedded in epon, contrasted, and cut in ultrathin slices for imaging with a Libra 120 transmission electron microscope (Zeiss, Oberkochen, Germany) as detailed in Supplementary material online, Methods.
2.7 Cardiac slices
Hearts from adult Balb/c mice were excised after cervical dislocation and immersed in oxygenated ice-cold preparation buffer (136 mM NaCl, 5.4 mM KCl, 1 mM MgHPO4, 0.9 mM CaCl2, 30 mM 2,3-Butanedionemonoxim, 5 mM HEPES, pH 7.4) and quickly embedded in low melting agarose (Carl Roth, Karlsruhe, Germany) at 37°C. Agarose blocks were kept in ice-cold preparation buffer supplemented with 10 mM glucose and cut into 300 µm thick coronary plane slices using a LeicaVT1200S vibratome. Slices were transferred onto 0.4 µm PTFE filter membranes (Millipore, Tullagreen, Ireland) and the filters were inserted in a 6-well cell-culture plate with 1.5 mL DMEM F12 medium (Life Technologies) supplemented with 20% knockout SR (Life Technologies), 1% MEM NEA (Life Technologies), 2 mM l-glutamin, 0.1% 2-mercaptoethanole, penicillin, and streptomycin. Slices were incubated at 37°C, 5% CO2, and 100% humidity. After application of either l-Tryp or l-Phen, slices were embedded in Jung Tissue Freezing Medium (Leica) and kryosectioned (HM500OM, Microm, Walldorf, Germany) into 10 µm thick samples that were used for immunostaining. Immunostaining was performed as described earlier with following exceptions: fixation was performed by 10 min heating to 37°C and for permeabilization 1% Triton X-100 in PBS was applied for 1 h.
2.8 Langendorff-heart preparation
Adult murine hearts were used for Langendorff isolated heart preparations on a commercially available apparatus according to manufacturer's instructions (ADInstruments, Spechbach, Germany). Details on the procedure are provided in Supplementary material online, Methods.
2.9 Statistics
Results are expressed as mean values ± standard error of mean (SEM). N signifies independent experiments, whereas n indicates independent samples. Statistical comparisons were performed using either Student's T-test for the comparison of two groups of samples (based on n) or one-way ANOVA together with Bonferroni's post-hoc test (based on n) in case of multiple group comparison or Dunnett's post-hoc test (based on N, statistics in Figure 5D), using Graph Pad Prism software. Significance was assumed for P ≤ 0.05.
3. Results
3.1 l-Tryp interfering with cadherin-tryptophan-swap disturbed Dsg2 localization and reduced cardiomyocyte cohesion in a dose-dependent manner
In order to evaluate the importance of certain ICD components, we first sought to influence general cadherin–cadherin interaction. One common binding mechanism among cadherins is the so called ‘tryptophan-swap’ in which a conserved tryptophan residue at position 2 of the first extracellular domain interacts with the cell-adhesion-recognition site of another cadherin.18 We used excess free l-Tryp to disturb this interaction as we showed before for Dsg3 binding.14 Application of l-Tryp in concentrations of 20 µM to 1 mM for 24 h together with physiological levels of Ca2+ was performed and the distribution of Dsg2 and N-Cad was assessed in HL-1 cardiomyocytes (Figure 1A). In control conditions, Dsg2 exhibited a finely dotted distribution primarily along the cell membrane of the HL-1 cells which partially colocalized with N-Cad staining. l-Tryp caused an accumulation of irregular Dsg2 streaks along the membrane of HL-1 cardiomyocytes. This effect seemed to be dose-dependent and was most obvious in cells treated with 1 mM l-Tryp. Interestingly, regular N-Cad staining along the membrane remained similar to controls. In order to rule out possible unspecific amino acid-related effects of l-Tryp, we applied another aromatic amino acid l-phenylalanine (l-Phen) in the same concentrations. l-Phen did neither change Dsg2 nor N-Cad staining patterns in HL-1 cells (Figure 1B). l-Tryp, but not l-Phen, seemed to affect primarily Dsg2 distribution in HL-1 cardiomyocytes, whereas N-Cad remained regularly distributed. Measurement of Dsg2-stained particle size with the ImageJ software corroborated the findings of immunofluorescence staining and revealed a slight but significant increase in size after treatment with l-Tryp or single peptide (SP). This desmoglein-specific SP consists of just one amino acid loop identical to the ones used in the TP.19 The peptide is predicted to bind to just one desmoglein molecule and therefore block the binding site to prevent trans-interaction with another desmoglein from a neighbouring cell. l-Phen or tandem peptide (TP) did not change the particle size (Figure 1C, N = 4–7 independent cell-culture passages, n = 6–14 wells). TP consists of two identical cyclic amino acid loops imitating part of the first extracellular domain of desmogleins and was shown to stabilize desmoglein but not classical cadherin trans-interaction by linking the desmoglein residue of one cell to the residue of another cell.19
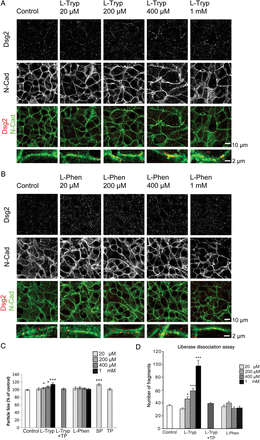
l-Tryp interfering with cadherin-tryptophan-swap disturbed Dsg2 but not N-Cad. (A) Inhibition of the cadherin-tryptophan-swap interaction by l-Tryp led to an accumulation of Dsg2 in condensed irregular streaks along the membrane of HL-1 cardiomyocytes (white arrows). This effect was dose-dependent and the membrane localization of the adherens junction protein N-Cad was not affected. Dsg2 and N-Cad immunostainings were partially colocalized at the membrane. (B) l-Phen served as a control treatment and did not change Dsg2 and N-Cad distribution. Scale bar = 10 µm or 2 µm. (C) Particle size of Dsg2 staining was quantitatively analysed using Image J software and showed a dose-dependent significantly increased streak size under l-Tryp treatment without effects of l-Phen. N = 5–11 independent cell-culture passages, n = 14–26 wells, *P ≤ 0.05; ***P ≤ 0.001 vs. PBS control. (D) Number of fragments per well after liberase-based cell dissociation. l-Tryp caused loss of cohesion in cardiomyocytes in a desmoglein-dependent manner. HL-1 cardiomyocyte monolayers were pretreated with l-Tryp, l-Phen, or l-Tryp with a desmoglein-specific TP (l-Tryp+TP) and subsequently subjected to a defined sheer stress, resulting in fragmentation; the number of fragments per well was quantified. l-Tryp-treated monolayers were less resistant towards defined sheer stress resulting in a dose-dependent increase of fragmentation, indicative of reduced cell–cell cohesion, whereas l-Phen did not exhibit this effect. l-Tryp-induced loss of cell–cell cohesion was prevented by co-incubation with a desmoglein-specific TP which strengthens Dsg2 trans-interaction. N = 4–7 independent cell-culture passages, n = 6–14 wells, *P ≤ 0.05; ***P ≤ 0.001 vs. PBS control.
Next, we determined the effects on cardiomyocyte cohesion under the same conditions. To accomplish this aim, we modified the dispase-assay approach, which is routinely used in keratinocytes,17 by supplementing dispase with liberase DH (a collagenase mixture for cardiomyocyte separation) as described in the Methods section. Control conditions yielded ∼35.6 ± 1.9 fragments. l-Tryp, but not l-Phen, in concentrations above 200 µM significantly increased the number of cell fragments after shear stress in HL-1 cardiomyocyte monolayers (Figure 1D, Supplementary material online, Figure S1). The inhibitory effect of l-Tryp on cardiomyocyte cohesion was abolished by co-incubation with a desmoglein-specific TP. These data indicate that l-Tryp interfered with cardiomyocyte cohesion primarily by impairing Dsg2-mediated binding.
3.2 Targeting of desmosomal adhesion revealed that Dsg2 is crucial for cardiomyocyte cohesion
We sought to corroborate specific desmoglein-mediated effects in desmosome redistribution further, by applying antagonistic SP. Application of SP (20 µM) to HL-1 cardiomyocyte monolayers led to a distribution pattern of Dsg2 similar to application of l-Tryp (400 µM) (Figure 2A). Dsg2 accumulated in irregular sized streaks along the cell membrane resembling the l-tryp effects. On the contrary, incubation with 20 µM TP resulted in a finely punctuated regular Dsg2 staining pattern similar to control conditions. The cyclic SP used in our experiments was modelled to correspond to the extracellular part of human Dsg1, however, also exhibits strong homology to the murine and human Dsg2 sequences (Figure 2D) and was shown to inhibit trans-interaction of Dsg3 also.14 TP corresponds to two cyclic SP entities which are linked to provide two cadherin binding sites.
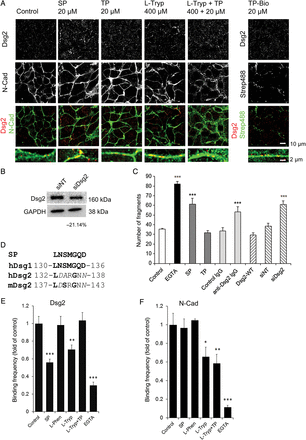
Specific targeting of Dsg2 interaction reduced cohesion in cardiomyocytes. (A) Immunostaining for Dsg2 and N-Cad showing that the inhibition of specific desmoglein–desmoglein interaction by a competing cyclic SP (20 µM, 24 h) led to Dsg2 condensation and irregular streaks along the membrane of HL-1 cardiomyocytes similar to the effects of l-Tryp (400 µM, 24 h). TP (20 µM, 24 h) changed the Dsg2 staining pattern from a finely punctuated distribution along the cell membrane to a more linear and regular fine staining. TP also ameliorated the effect of l-Tryp (l-Tryp + TP, 400 µM, 24 h). Biotinylated TP (Bio-TP, 20 µM, 24 h) accumulated at the membrane as shown by streptavidin-AlexaFuor488 (Strep488) and was partially colocalized with Dsg2. Neither of the conditions tested affected N-Cad localization along the cell membrane. Scale bar = 10 µm or 2 µm. (B) Dsg2 was confirmed to be down regulated by 21.14% ± 9.42% in HL-1 cells transfected with siRNA directed against murine Dsg2 using western blot, N = 4 independent cell-culture passages. (C) Calcium chelation by EGTA (5 mM, 1 h) resulted in a remarkable monolayer fragmentation in the liberase-based cell dissociation assay. Treatment of HL-1 cells with SP (200 µM, 24 h) competing for desmoglein-interaction significantly increased monolayer fragmentation in the liberase assay, whereas TP (400 µM, 24 h) did not change the number of fragments after shear stress. An antibody directed against the extracellular part of Dsg2 (anti-Dsg2-IgG, 2.5 µg/mL, 24 h) was able to compete for cell–cell cohesion, whereas an antibody against the intracellular part of Dsg2 (control a-Dsg2, 2.5 µg/mL, 24 h) had no effect. Overexpression of Dsg2 decreased monolayer fragmentation. Knockdown of Dsg2 (siDsg2) weakened cell–cell cohesion and increased fragmentation, whereas irrelevant non-target siRNA (siNT) had no effect. Control conditions consisted of PBS-treated HL-1 cells. ***P ≤ 0.001 vs. PBS control, N = 3–11 independent cell-culture passages, n = 6–22 wells. (D) Comparison of the SP structure with the respective loci in human (h) Dsg1 (NCBI Reference Sequenze NP_001933.2) and Dsg2 (NCBI GeneBank AAH99656.1) and murine (m) Dsg2 (NCBI GeneBank BAB86843.1). Identical amino acids are marked in bold and similar amino acids in italics in the standard one-letter code. (E) Atomic force microscopy measurement of two independent coatings (N = 2) of purified human Dsg2 revealed that SP (20 µM) and l-Tryp (400 µM) reduced homophilic binding frequency, whereas l-Phen (400 µM) had no effect. TP (20 µM) rescued the disrupting effect of l-Tryp and EGTA (5 mM) served as a control which strongly reduced Ca2+-dependent Dsg2 binding. n = 4–6 independent measurements. **P ≤ 0.01; ***P ≤ 0.001. (F) Homophilic binding of two indepedent coatings of purified mouse N-Cad was not affected by SP (20 µM), or l-Phen (400 µM). l-Tryp (400 µM) reduced binding in N-Cad similar to Dsg2, however, the effect on N-Cad was not blocked by TP. The binding frequency was maximally reduced by EGTA (5 mM). n = 4–6 independent measurements, *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Co-incubation of TP ameliorated the effects of 400 µM l-Tryp and led to a Dsg2 staining pattern with fewer and slightly smaller Dsg2-positive dots (Figure 2A, Figure 1C). Biotin-labelling of TP (TP-Bio) revealed partial colocalization with Dsg2. Overall, the distribution pattern more closely resembled the punctuated localization of Dsg2 rather than the continuous distribution of N-Cad along cell junctions. N-Cad staining was not affected by SP, TP, l-Tryp, or l-Tryp+TP (Figure 2A).
In the next part of our study, we analysed Dsg2-mediated cardiomyocyte cohesion specifically (Figure 2C, N = 3–11 independent cell-culture passages, n = 6–22 wells). Similar to Ca2+-depletion by EGTA, which reduced overall cadherin trans-interaction because Ca2+ is required to rigidify the cadherin extracellular domain, specific desmoglein-targeting by SP led to almost twice the number of fragments when compared with control. TP did not significantly alter fragment number. An antibody directed against the extracellular part of murine Dsg2 (anti-Dsg2-IgG) significantly inhibited cardiomyocyte cohesion. In contrast, a control antibody directed against the intracellular loop of Dsg2 (control IgG) had no effect. Overexpression of human wild-type Dsg2 (Dsg2-WT, Figure 4) slightly decreased the number of fragments indicating enhanced cardiomyocyte cohesion. In contrast, siRNA-induced reduction of the endogenous murine Dsg2 (Figure 2B) destabilized cell–cell cohesion significantly, whereas non-targeting siRNA had no effect compared with control.
In order to link the effects observed in HL-1 cells to reduced Dsg2 interaction, we performed atomic force microscopy binding studies using the extracellular parts of Dsg2 (Figure 2E, N = 2 independent coatings, n = 4–6 independent measurements) or N-Cad (Figure 2F, N = 2 independent coatings, n = 4–6 independent measurements), respectively. In contrast to l-Phen treatment, l-Tryp disturbed both homophilic Dsg2 and N-Cad interaction. The inhibitory effect of l-Tryp was abrogated by co-incubation with TP in Dsg2, but not N-Cad, binding. SP reduced homophilic Dsg2 but not N-Cad trans-interaction, demonstrating a Dsg2-specific effect of SP and TP. EGTA treatment largely abolished both Dsg2 and N-Cad binding.
3.3 Ultrastructural changes of ICD organization in response to L-Tryp and SP
Electron microscopy imaging of adherent confluent HL-1 cells revealed desmosome-like areae compositae with typical electron dense plaques (Figure 3A), as well as adherens junction-like areae compositae (Figure 3B, N = 2 independent cell-culture passages, n = 161–265 junctions) and mixtures of both cell contact types in all conditions tested. However, l-Tryp (400 µM, 24 h) as well as SP (20 µM, 24 h) significantly increased adherens junction-like but not desmosome-like area composita length and led to irregularities and disruptions within these long junctions (Figure 3A and B, arrow heads). Irregular and disrupted areae compositae were 2.4 times more frequent in l-Tryp treated cells compared with either PBS or l-Phen (400 µM, 24 h). Similarly, SP-treated cells showed a 1.5 fold increase in irregular and disrupted junctions.
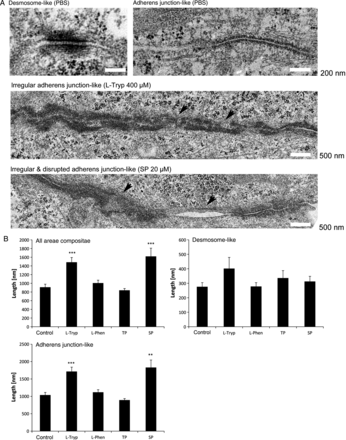
Interference with Dsg2 binding was associated with ultrastructural alterations of areae compositae. (A) Electron microscopy images of the ultrastructure of HL-1 cells revealed desmosome-like areae compositae with broader electron dense plaques than in the less prominent areae compositae under control conditions (PBS). Treatment with l-Tryp (400 µM, 24 h) or SP (20 µM, 24 h) led to longer adherens junction-like areae compositae with frequent irregularities and disruptions (arrows). (B) Statistical analysis of 161 to 265 adhesive junctions per condition in two independent experiments showed a significant increase in the mean area composita length (length of desmosome-like connections + length of adherens junction-like connections divided by the number of connections) under l-Tryp and SP treatment, whereas TP (20 µM, 24 h) or l-Phen (400 µM, 24 h) had no effect. The increase in length was caused by an increase of adherens junction- rather than desmosome-like area composita length. **P ≤ 0.01; ***P ≤ 0.001 vs. PBS control.
3.4 Certain Dsg2 mutations caused loss of cardiomyocyte cohesion
Dsg2 mutations underlie AC in ∼10% of all patients. We tested selected Dsg2 mutations in our liberase assay. Current theories predict that one mutated allele of a desmosomal gene could lead to production of a dominant negative protein which integrates into desmosomes but cannot perform regularly and therefore weakens the desmosomal cohesion. We expressed human native Dsg2 (WT), as well as nine Dsg2 variants with point mutations of the following amino acids: D154E, D264E, N266S, D494N, A517V, G812S, G812C, C813R, and V920G. All constructs showed membrane localization in transfected HL-1 cardiomyocytes as evaluated by detection of EGFP fluorescence during confocal microscopy (Figure 4A). We detected a band of ∼180 kDa corresponding to the human Dsg2 construct with its EGFP-tag in western blot analysis (Figure 4B). This band was not present in HL-1 cells transfected with the empty pEGFP-N1 vector (GFP). Endogenous murine Dsg2 migrated at ∼160 kDa and α-tubulin migrating at 55 kDa was used as loading control. Our functional liberase-assay revealed that of the Dsg2 mutations tested, only the A517V and the V920G mutations led to an increase in fragmentation upon shear stress and therefore a reduction of cell–cell cohesion in HL-1 cardiomyocytes. Expression of wild-type human Dsg2 slightly reduced fragmentation as shown earlier, whereas the mutations D154E, D264E, N266S, D494N, G812S, G812C, and C813R did not influence cardiomyocyte cohesion (Figure 4C, N = 6–11 independent cell-culture passages, n = 10–18 wells).
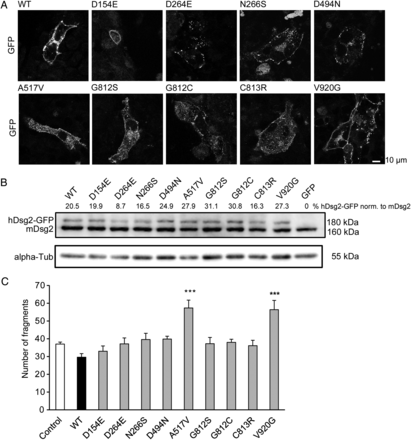
Certain Dsg2 mutations led to loss of cardiomyocyte cohesion. (A) Mutated versions of human Dsg2 expressed in HL-1 cardiomyocytes showed membrane location as assessed with the attached GFP-tag using confocal fluorescence microscopy. Scale bar = 10 µm. (B) HL-1 cardiomyocytes transfected with these Dsg2 mutant versions displayed two anti-Dsg2-antibody-detectable bands in western blot analysis. One migrated at 160 kDa molecular weight and corresponded to the endogenous murine Dsg2 protein, the second migrated at 180 kDa and corresponded to the human Dsg2 mutant which owes its higher molecular weight to the attached GFP-tag. The latter was undetectable in HL-1 cells transfected with the Dsg2-free GFP-vector. The expression level of the human Dsg2 mutants was ∼25% of the expression level of endogenous murine Dsg2. alpha-Tubulin (55 kDa) was used as a loading control. (C) Expression of the native human Dsg2 protein in HL-1 cardiomyocytes (WT) led to a slight protection from monolayer fragmentation in the liberase assay, whereas expression of either mutant A517V or V920G cause an increase of monolayer fragmentation indicative of loss of cell–cell cohesion. The other Dsg2 mutants tested did not significantly alter fragmentation. ***P ≤ 0.001 vs. PBS control. N = 6–11 independent cell-culture passages, n = 10–18 wells.
3.5 l-Tryp- and SP-mediated interference with desmoglein-binding altered cardiomyocyte function and dampened the adrenergic response of intact adult mouse hearts
To bring our study on cardiomyocyte cohesion one level closer to the actual (patho)physiological setting, we performed functional tests on isolated Langendorff-perfused adult murine hearts. The experimental protocol included a 10 min baseline evaluation followed by 15 min of 2 mM l-Tryp/l-Phen or 20 min of 40 µM SP perfusion. Then, we washed out the respective reagent for 10 min and applied a strong adrenergic stimulus by infusing 2 µM Iso for 10 min and ended the experiment with a 15 min washout. The Langendorff perfusion was performed in constant pressure mode and therefore the coronary flow of the heart determined how quickly an applied agent reached the heart in our setting and how quickly it was washed out again. We could optically monitor this process by colouring l-Tryp, l-Phen, or Iso with phenol red. During the measurements, we evaluated the heart rate and the pressure changes in the aortic cannula. Pulse pressure per heart beat (Figure 5A), heart rate (Figure 5B), and the rate-pressure-product (RPP, Figure 5C) were calculated as means of 10 s intervals and compared with each other (Figure 5D, N = 9–11 mouse hearts per condition). Neither application of l-Tryp, SP, or l-Phen significantly changed the pulse pressure of the perfused hearts, even though l-Tryp application led to a slight but insignificant reduction. However, SP showed a slight decrease of pulse pressure during perfusion and subsequent washout. Application of high-dose Iso led to the expected transient positive inotropic effect in hearts. l-Tryp and SP, but not l-Phen, reduced heart rate during acute infiltration. Subsequent Iso application resulted in an increase in heart rate in l-Phen- and less so in l-Tryp-pretreated hearts. Iso application led to a huge fluctuation in heart rate from ∼80–340 bpm in l-Tryp- or SP-pretreated cells. Interestingly, in SP-pretreated hearts, however, the positive chronotropic effect of Iso was gone. RPP indicates a slight reduction of heart work during l-Tryp treatment which was more pronounced during SP, but not observed in response to l-Phen application. This was followed by a huge increase in RPP upon the first minute of Iso-challenge in l-Phen- and l-Tryp-, but less so in SP-pretreated hearts. These data showed on the organ level that interference with desmosomal binding impaired the hearts' ability to adequately respond to adrenergic challenging.
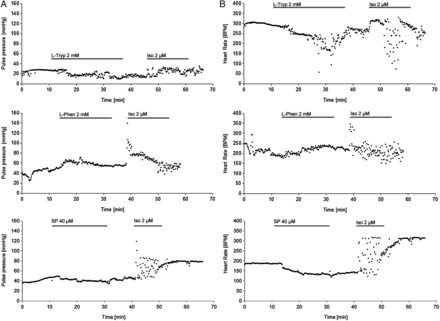
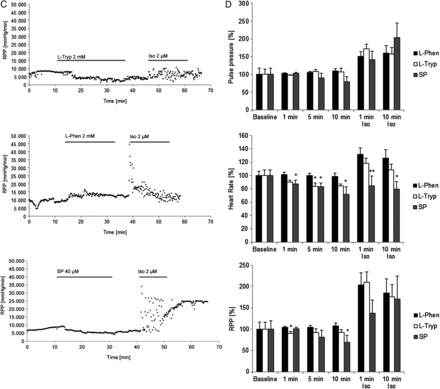
Interference with desmoglein binding impaired cardiomyocyte function and dampened the adrenergic response of intact adult mouse hearts. (A) The pulse pressure in the aortic cannula developed by the mouse hearts was not affected by either l-Tryp (2 mM) or l-Phen (2 mM) added to the perfusion buffer. However, it increased acutely in the first minutes of isoproterenol (Iso, 2 µM) application. (B) l-Tryp as well as SP treatment induced irregularity and a drop in heart rate, whereas l-Phen treatment did not cause such an effect. After Iso application there was an adequate acute positive chronotropic effect in l-Phen pretreated hearts, however, this effect is significantly reduced in SP- (and to a lesser degree in l-Tryp-) challenged preparations, where Iso only caused further irregularities in heart rate measurements. (C) The rate-pressure-product (RPP, pulse pressure multiplied with heart rate) indicates a drop in heart work after SP (and to a lesser degree l-Tryp) application, Iso following this treatment led to a fluctuation in RPP, which summed up to an overall slight reduction of RPP after Iso in SP-pretreated hearts. In contrast, l-Phen did not influence RPP and subsequent Iso infiltration led to a sharp increase in RPP. (D) Bar graphs represent mean data of N = 9–11 mouse hearts per condition. Non-repeated one-way ANOVA analysis with Dunnett's post-hoc test was performed at each time point indicated, *P ≤ 0.05; **P ≤ 0.01 vs. l-Phen.
To investigate the mechanism underlying the reduced adrenergic response, we employed an ex vivo technique using adult murine cardiac slices. The advantage of this technique is that it is possible to harvest several slices from a single heart and use them for different treatment conditions. l-Tryp (1 mM, 1 h) caused a rapid opening and tearing of ICDs which was demonstrated by immunostaining against N-Cad and beta1-adrenergic receptor (Figure 6A). Moreover, as reported in the literature under conditions of impaired cardiomyocyte cohesion,20 we also found lateralization of connexin 43 immunostaining to areas outside the ICDs upon l-Tryp treatment (Figure 6B).
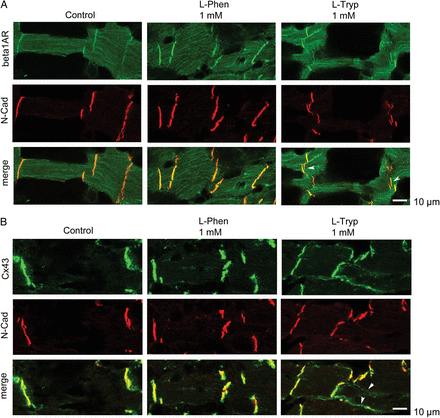
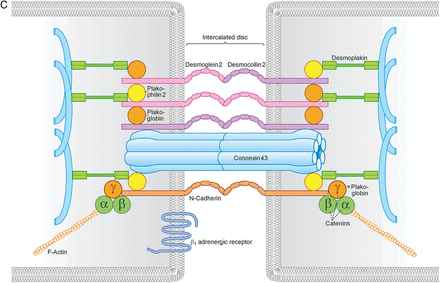
l-Tryp interfered with ICD morphology and lateralized connexin 43. (A) l-Tryp (1 mM, 1 h) opened up ICDs in intact cardiomyocytes within a murine cardiac slice. Under control conditions as well as with l-Phen (1 mM, 1 h) both N-Cad and beta1-adrenergic receptor immunostainings were localized within the ICD as a single band at the short end of cardiomyocytes. l-Tryp interfered with ICD morphology and opened up the cardiomyocyte cell–cell contacts leading to a double-band staining pattern (arrows). Scale bar = 10 µm. (B) l-Tryp caused marked lateralization of connexin 43 staining (arrows), which is restricted to the intercalated disc in control and l-Phen conditions. Scale bar = 10 µm. The pictures show representative results from N = 7–9 independent heart slices and n = 17–20 independent staining procedures. (C) Schematic representation of ICD components.
4. Discussion
In this study, we showed that inhibition of Dsg2 interaction led to changes in the distribution of Dsg2 in HL-1 cardiomyocytes and reduced cardiomyocyte cohesion in a functional dissociation assay. Furthermore, we demonstrated that certain Dsg2 mutations implicated in AC reduce cardiomyocyte cohesion, whereas other Dsg2 mutations do not work via this mechanism. On the organ level, interference with desmoglein trans-interaction reduced heart rate, cardiac work, and also dampened subsequent response to adrenergic challenging in Langendorff-mouse-heart preparations.
l-Tryp treatment is known to inhibit Dsg trans-interaction as shown for Dsg3 by single-molecule atomic force measurements,14 presumably by competing with the cadherin-tryptophan-swap mechanism. In immunostaining of l-Tryp-treated HL-1 cells, we detected irregular condensation of Dsg2, but not of N-Cad staining. In line with this, l-Tryp decreased cardiomyocyte cohesion in the dissociation assay in a dose-dependent manner and these effects were prevented by the Dsg-specific TP. TP application was previously shown to strengthen desmoglein trans-interactions,19 however, it did not increase cardiomyocyte cohesion above control levels. Specific inhibition of desmoglein-interaction by SP resulted in Dsg2 staining patterns similar to l-Tryp-treated cells without effects on N-Cad staining. Atomic force microscopy revealed that l-Tryp targets both Dsg2 and N-Cad homophilic interactions, whereas SP and TP were found to be Dsg2-specific and did not affect N-Cad binding. Taken together, these data indicate that l-Tryp, under the conditions used in this study, reduced cardiomyocyte cohesion primarily by targeting Dsg2. Specific approaches targeting Dsg2 with inhibitory antibody or siRNA also reduced HL-1 cohesion further indicating that Dsg2 is crucial for cardiomyocyte cohesion. These data are in agreement with results in other cell lines expressing Dsg2 as the only desmoglein isoform. In enterocytes, Dsg2-mediated binding is crucial for cell cohesion and intestinal epithelial barrier function.21,22 In contrast, in keratinocytes, which express several Dsg isoforms, the relevance of Dsg2 for cell cohesion was less pronounced than that of Dsg3. Interestingly, siRNA-mediated depletion of Dsg2 caused cell sheet fragmentation only under conditions of increased shear.22
The inhibitory effects of l-Tryp and SP were paralleled by ultrastructural alterations of adherens junction-like areae compositae, whereas desmosome-like areae compositae were not affected significantly. We found that both l-Tryp and SP increased adherens junction-like area composita length which was accompanied by plaque irregularities and junction disruption.
After we established that l-Tryp and more specifically SP can be used to target Dsg2-mediated cardiomyocyte cohesion, we applied this approach on the organ level using the Langendorff-model. The acute reduction of heart rate and RPP in response to l-Tryp and SP as well as the dampening of the adrenergic response after SP-challenging in the murine Langendorff-model may be a direct effect of reduced Dsg2 interaction causing impaired force generation. Alternatively, the reduction of heart work as evaluated by RPP might be caused by impaired conduction due to gap junction dysfunction, which is suggested by the relocalization of connexin 43 outside the ICD upon l-Tryp treatment in murine cardiac slices. Connexin 43 lateralization in the context of AC has been described previously.20 It is also possible that the cardiac desmosomal components might influence functionality of the β-receptor system directly (Figure 6C). This is indicated by our observation that disrupted ICD following l-Tryp treatment were accompanied by disrupted beta1-adrenergic receptor staining at the ICD. The mechanisms of this possible interaction will have to be evaluated in further studies.
In addition to our experiments characterizing the role of Dsg2 for cardiomyocyte cohesion and function, we also employed a dissociation assay to investigate the effects of Dsg2 mutations associated with AC on cardiomyocyte cohesion. Expression of the native human Dsg2 protein strengthened cardiomyocyte cohesion proving that the dissociation assay is not only valuable for detecting agents that cause fragmentation but also for such conditions that prevent fragmentation and thus may be useful to test new therapeutic strategies in the future. Although we expressed human native Dsg2 in a murine cell line, we can assume by its localization and functional cohesion strengthening that the heterologous protein is integrated into mouse area compositae in HL-1 cells. As previously shown for the Dsg2 mutants G812S, G812C, and C813R, in neonatal rat cardiomyocytes, the mutants used in this work were also expressed at the cell membrane in HL-1 cardiomyocytes.8 Protein expression levels of the respective Dsg2 mutants were comparable, precluding a transcriptional effect of the mutation. In our functional assay, the Dsg2 mutations A517V and V920G caused loss of cardiomyocyte cohesion, suggestive of a dominant negative effect. A517V lies in the anchor domain and V920G in the intracellular repeated unit domain of Dsg2, therefore it is unlikely that these mutations affect the extracellular domain-binding mechanism directly. The A517V mutation has been identified in an AC patient (with an additional Dsc2 mutation) and did not change Dsg2/Dsc2-binding properties or PG membrane location.23 A517V and V920G are classified as proteins of unknown pathogenicity,4 and V920G has been found in 0.8% of the healthy population.24 The fact that in our assay these two mutations cause loss of cohesion indicates that they can, under certain conditions, reduce Dsg2 binding. Moreover, this observation suggests that in cultured cells in vitro overexpression of a single mutant Dsg2 isoform can be more devastating than in patients. The remaining Dsg2 mutants tested did not decrease cardiomyocyte cohesion in comparison to control conditions. However, unlike expression of native Dsg2, they also did not strengthen cell cohesion either. D154E,25 D264E,26 and N266S27 are located in the first two extracellular domains and were shown to be either pathogenic or of unknown significance. Since in our study they did not interfere with cardiomyocyte cohesion significantly, these data suggest that these mutations may contribute to AC pathogenesis by other mechanisms such as modulation of cell signalling as has been proposed previously.28
5. Conclusion
Here we employed a functional assay for cardiomyocyte cohesion, which demonstrated decreased cohesion under conditions impairing Dsg2 interaction. Interestingly, Dsg2 appears to be more relevant for cardiomyocyte cohesion than N-Cad. Two Dsg2 mutations associated with AC decreased cardiomyocyte cohesion, whereas 7 different mutations are pathogenic due to other mechanisms. Moreover, we showed that inhibition of cadherin-interaction in the heart led to impaired heart rate, RPP, and blunted reaction to adrenergic stimulation. More research on the interaction of area composita and β-adrenergic receptors especially in the context of AC and other forms of cardiomyopathy is warranted.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Friedrich-Baur-Stiftung (grant number 01/12 to A.S.).
Acknowledgement
We thank Angelika Antonius, Martina Hitzenbichler, Sabine Schäfer, Sabine Mühlsiemer, and Cathleen Plietz for expert technical assistance, Michael Christof for design of Figure 6C, Mariya Radeva for statistical advice, Forian Brettner for the perfusion pump, and Prof. William Claycomb for providing the HL-1 cardiomyocytes.
Conflict of interest: none declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}