





Abstract
Vascular permeability is essential for the health of normal tissues and is an important characteristic of many disease states. The role of the Wnt/frizzled pathway in vascular biology has recently been reported. The objectives of this study are to analyse the role of Frizzled7 (Fzd7) receptor in the control of vascular integrity.
Fzd7 is expressed in endothelial cells and accumulates at the points of cell–cell contact in association with VE-cadherin and β-catenin, two major adherens junction molecules. To selectively delete fzd7 in the vasculature, we developed gene targeting approaches using CreLox strategy in mice. Genetic fzd7 inhibition in the endothelium increases vascular permeability in basal and factor-induced conditions. On the cellular level, fzd7 knockdown or depletion leads to an increase in paracellular permeability with a loss of adherens junction organization. These impairments are associated with a decrease in both VE-Cadherin and β-catenin expression, a decrease in their association and an increase of tyrosine phosphorylation of VE-cadherin/β-catenin. Fzd7 transduces a Wnt/β-catenin signalling cascade that is required to regulate β-catenin and canonical target gene expression. Finally, LiCl, a GSK3 inhibitor, and β-catenin overexpression rescued endothelial integrity and adherens junction organization, induced by fzd7 deletion.
These findings establish that Fzd7 is a new partner of adherens junctional complex and represents a novel molecular switch for the control of vascular permeability via activation of the Wnt-canonical pathway.
1. Introduction
The function of the endothelium is to provide a network to allow delivery of oxygen and nutrients to tissues throughout the body. This network comprises adjacent endothelial cells which utilize adherens junction complexes containing vascular endothelial cadherin (VE-cadherin) link to catenin proteins (α, β, γ or p120) to maintain the appropriate level of vascular permeability.1,2 In a variety of pathological conditions including ischaemic disease, acute and chronic inflammation, and tumour formation, secreted mediators lead to a prolonged and chronic increase in endothelial paracellular permeability which can result in vessel wall leakiness and organ dysfunction.3
The Wnt/Fzd pathway is known to play important roles in multiple physiological and pathological processes.4 Recent advances in vascular biology highlight important roles for multiple components of the Wnt/Fzd signalling pathway in endothelial cell biology regulating cell differentiation, proliferation, survival, cell junctions, and polarity.5,6 Wnt/Fzd signalling is a complex pathway with 19 soluble Wnt proteins that can bind to 10 frizzled receptors and activate canonical, dependant to the β-catenin, and non-canonical signalling pathways, so-called planar cell polarity (PCP) or the Wnt/Ca2+ pathway.7 Among the Fzd receptors, we and others have previously demonstrated that Frizzled7 (Fzd7) is expressed in EC and that its down-regulation enhances EC spreading and modifies actin cytoskeleton.8,9 Different reports have demonstrated the role of Fzd7 in development,10 stem cell renewal and differentiation,11 and cancer progression.12 However, its role in the vasculature has not yet been fully elucidated.
In the current study, we demonstrate that Fzd7 is a constituent of adherens junctions in endothelial cells. To study specific Fzd7 endothelial functions, we generated mice with an endothelium-restricted deletion of the fzd7 gene using a Cre/loxP system (fzd7ECKO). Deletion of fzd7 in EC, induced an increase in endothelial leakage in physiological condition, after VEGF activation and inflammatory conditions. We then provide evidence that Fzd7 signalling regulates paracellular permeability, junction organization, and VE-cadherin/β-catenin expression. Fzd7 activates the canonical Wnt pathway which is able to control gene expression and cadherin-mediated cell–cell junctions through the regulation of β-catenin and VE-cadherin expression levels.
2. Methods
2.1 Mice
Homozygote floxed fzd7floxP/floxP mice were generated and were crossed with Tie2-driven Cre-recombinase mice (Tie2-Cre) to generate fzd7 knockout mice in the endothelium (fzd7ECKO).
2.2 Miles assay in mice
Mice were intravenously injected with Evans Blue (100 µL) in the tail vein. After 30 min, mice were anaesthetized with 1.5% isoflurane and 100 µL of PBS or VEGF (20 ng) was injected sub-dermally. After 30 min, mice were euthanized by overdose of pentobarbital intraperitoneally and skin samples dissected and placed in formamide at 56°C to extract Evans Blue for 24 h. The absorbance of extracted dye was measured at 630 nm.
2.3 Model of chronic cutaneous irritation
5% SDS/PBS was administrated epicutaneously once per day for 10 days as described by Cramer et al.13 Mice were euthanized by overdose of pentobarbital intraperitoneally. Skin samples were harvested, fixed in 4% PFA, and mounted in paraffin. Haematoxilin and eosin staining was performed to quantify thickness.
2.4 Cell culture, transfection, and growth factor activation
Human umbilical vein endothelial cells (HUVEC), MS1 endothelial cells, and HEK-293 cells were cultured as previously described.8,14 Small interference siRNA-mediated knockdown was performed using Interferin (Polyplus) following the manufacturer's protocol. LiCl activation (Sigma) was performed in serum-free media at 30 mM. HUVEC were transduced with lentiviral vector at 10 MOI endoding for β-catenin (pTRIP-β-cat, plasmid from MA Buendia, Institut Paster, France15) or for eGFP as control.
2.5 Endothelial cell isolated from mice
Mice were sacrificed by inhalation of a lethal dose of CO2. Primary EC from WT and fzd7ECKO mice were isolated from kidneys or lung after magnetic separation with MS MACS columns (Miltenyi) as described previously.14
2.6 Immunofluorescence staining
Immunostaining on cells was performed as previously described.14,16 The following antibodies were used: hVE-cadherin (Santa Cruz), mVE-cadherin (Pharmingen), β-catenin (Sigma), active β-catenin (Upstate), Myc (Upstate), α-tubulin (Sigma), and mCD31 (BMA).
2.7 Immunoprecipitation
Plasmid constructions used for immunoprecipitation obtained from other laboratories included: pcDNA3.1(−)dsRed VE-cadherin (courtesy of Dr D. Gulino-Debrac, University of Grenoble, France),17 pCRD Fzd7 myc GPI (kindly provided by Dr Jeremy Nathans, Johns Hopkins University, Baltimore),18 and pmFzd7-myc (courtesy of Dr M. Moncouquiol, Magendie Institute, Bordeaux, France). pcDNA3.1(−) ΔCRD mFzd7-myc::his was produced in our laboratory. HEK-293 cells were transfected by using Lipofectamine (Invitrogen). Immunoprecipitation analyses were performed as previously described.14 Anti-Myc (Euromedex, clone 4A6), anti-Cad3 antibody (generously provided by D. Gulino-Debrac, University of Grenoble, France),17 anti-phospho Tyr (4G10) were used for immunoprecipitation assays.
2.8 Western blotting
Western blot was performed as previously described.8,14 Membranes were incubated with the following antibodies: α-tubulin (Sigma), Myc (Millipore), VE-cadherin (Santa Cruz), β-catenin (Sigma), active β-catenin (Upstate), total GSK3β (Euromedex), phospho-GSK3β (Ser9) (Cell signaling).
2.9 Paracellular tracer flux analysis
HUVECs, MS1, and EC isolated from mice were seeded on 0.4 µm pore size inserts (BD Biosciences), cultured in culture medium and assayed for permeability to FITC-dextran (40 kDa) as previously described.19
2.10 Analysis of mRNA expression by quantitative reverse transcriptase–polymerase chain reaction
Quantitative reverse transcriptase–polymerase chain reaction (qRT-PCR) analysis was performed as previously described.14 All experiments were performed in triplicate and differences in cDNA input were compensated by normalization to expression of β-actin or GAPDH.
2.11 Reporter gene assay
HUVEC were transiently transfected with 0.2 ng of 8XTOPflash reporter, 0.1 ng of β-galactosidase reporter, and various siRNA as indicated for individual experiments. Cells were co-cultured with CHO cells transfected with a plasmid harbouring β-gal (control), Wnt1, or Wnt5A. Wnt3A CM was obtained from cell lines expressing wnt3A (L Wnt3A CRL 2647) as described in the American Type Culture Collection (ATCC) protocol. Luciferase activity was determined as previously described.14
2.12 Statistical analysis
Statistical analysis was performed with Prism software (GraphPad). Results are reported as mean ± standard error of the mean (SEM). Comparisons between groups were analysed for significance with the non-parametric Mann–Whitney test. Differences between groups were considered to be significant when P≤ 0.05.
3. Results
3.1 Fzd7 is expressed in EC junctions and interacts with VE-cadherin and β-catenin
Clusters of differentiating mouse embryonic stem (ES) cells, designated embryoid bodies (EB) faithfully mimic vascular development in an in vivo-like fashion.20 We took advantage of embryoid bodies (EB) model to investigate the protein expression and localization in endothelial cells.20 We first studied its expression pattern following VEGF-induced EC differentiation. Immunostaining revealed Fzd7 expression during EB differentiation, first in a cluster pattern, then in the centre of the EB core, and ultimately throughout the core (see Supplementary material online, FigSupplementary DataA). At this late stage, co-staining with VE-cadherin demonstrated expression of Fzd7 in VE-cadherin positive cells and a co-localization at points of cell–cell contact (Figure 1A).
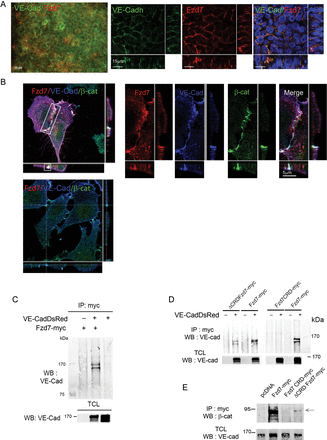
Fzd7 is expressed in EC cell–cell junctions, and co-localizes and interacts with VE-cadherin/β-catenin adherens junction complexes. (A) EB cultured with VEGF for 10 days were co-stained with Fzd7 (red) and VE-cadherin (green), and analysed with epifluorescence and confocal microscopy. (B) HUVEC were transfected with plasmid encoding for Fzd7-myc (top panel) or empty plasmid (bottom panel) and triple stained for myc (Fzd7, red), VE-cadherin (VE-Cad, Blue), and β-catenin (β-cat, green). High magnification of cell–cell junction in HUVEC transfected with Fzd7-myc plasmid; images are merged to visualize the co-localization at points of endothelial cell–cell contact (merge). (C) HEK-293 cells were co-transfected with empty plasmid (−) or a plasmid encoding for Fzd7-myc (+) and with (+) or without (−) plasmid encoding for VE-Cad-DsRed. Cell lysates were immunoprecipitated with anti-myc (IP : myc) and probed with anti-VE-cad antibody. Western blot (WB) with anti-VE-cad in total cell lysates (TCL) was performed to confirm VE-cadDsRed overexpression (PM ∼170 kDa). (D) HEK were co-transfected with different Fzd7 mutants and with (+) or without (−) VE-Cad-DsRed. Cell lysates were immunoprecipitated with anti-myc, followed by WB with anti-VE-cad antibody. WB with anti-VE-cad in total cell lysates (TCL) was performed to confirm VE-Cad-DsRed overexpression. (E) HEK-293 cells were co-transfected with VE-Cad-DsRed and fzd7 mutants. Cell lysates were immunoprecipitated with anti-myc and probed with anti-β-catenin antibody (PM 92–95 kDa). WB with anti-VE-cad in total cell lysates (TCL) was performed to confirm VE-cadDsRed overexpression (PM ∼170 kDa).
In order to investigate whether Fzd7 could be a new component of endothelial cell adheren junctions, we further examine sub-cellular distribution of the three partners Fzd7, VE-cadherin, and β-catenin. HUVEC transfected with full length Fzd7-myc plasmid (Figure 1B, top panel) or empty plasmid (Figure 1B, bottom panel) were immunostained with VE-cadherin, β-catenin, and myc antibodies (Figure 1B). We observed that Fzd7-myc strongly co-localizes with endogenous VE-cadherin and β-catenin complexes at points of cell–cell contact (Figure 1B, top panel, and high magnification). To quantitate the degree of co-localization between Fzd7/VE-cad and Fzd7/β-cat, analysis of micrographs of triple immunofluorescent labelling was performed. A Pearson coefficient for co-localization of full length Fzd7-myc/VE-cad was 0.53 ± 0.004 and 0.47 ± 0.04 for β-catenin (n = 10 images, from three independent experiments). For comparison, this coefficient is ∼0.45 for VE-cadherin and β-catenin staining, which are known to form junctional cell–cell complexes. Together these data demonstrate that Fzd7 is strongly expressed at points of cell–cell contact and suggest that Fzd7 may be part of VE-cadherin/β-catenin complexes at EC junctions.
We then evaluated potential molecular interactions between Fzd7 and VE-cadherin. Using HEK293 cells co-transfected with Fzd7-myc and VE-CadherinDsRed, we found that the anti-myc immunoprecipitate could bring down VE-cadherin (Figure 1C). We next identified the domains of Fzd7 required for interaction with VE-cadherin. Fzd7 mutants either with the extracellular domains which contain the cystein rich extracellular domain (CRD) linked to the GPI anchor (Fzd7CRD-myc) or Fzd7 deleted of the extracellular domain (ΔCRD Fzd7-myc) (see Supplementary material online,Supplementary DataB and C) were co-transfected with VE-cadherinDsRed. As shown in Figure 1D, ΔCRD Fzd7-myc brought down VE-cadherin, whereas Fzd7CRD-myc did not, suggesting that Fzd7/VE-cadherin interaction is not mediated by the Fzd7 extracellular domain but involves its intracellular domain. Furthermore, western blot analysis revealed that β-catenin was detected in the myc immoprecipitate (Figure 1E). Collectively, these results suggest that Fzd7 can interact directly to form a multi-protein complex with VE-cadherin and β-catenin. These data support the hypothesis that Fzd7 may play a role in the regulation of cell–cell adhesion.
3.2 In vivofzd7 deletion increases vascular permeability
RT–PCR performed on cells isolated from the lung tissue from mice demonstrated that fzd7 transcripts were not restricted to EC and were expressed at a consistent level in CD31+ cells (Figure 2A). To study specific Fzd7 endothelial functions, we generated mice with an endothelium-restricted deletion of the fzd7 gene using a Tie2-Cre/loxP system (Figure 2B).21 Functionality of the model was verified by β-galactosidase (β-gal) staining in tissues of mice crossed onto a Rosa26-Stopfl-LacZ reporter background (Figure 2C). EC-specific β-gal staining was detected in EC only in Cre-Tie2 mice (Figure 2C, b–e). Quantitative PCR analysis showed a drastic reduction in fzd7 mRNA expression in EC rich tissues such as the lung from fzd7ECKO compared with WT littermate mice (Figure 2D). Moreover, EC isolated from WT and fzd7ECKO mice demonstrated the specific deletion of fzd7 in EC (Figure 2D). Fzd7ECKO mice were viable without gross alteration or phenotype during the first 3 months of life.
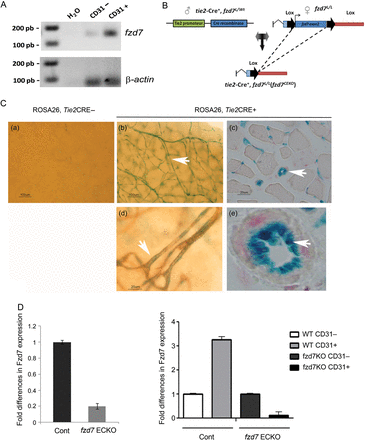
Generation of fzd7ECKO mice. (A) fzd7 transcript analysis after RT–PCR on EC isolated from mouse lung in CD31 negative (CD31−) and CD31 positive (CD31+) enriched fractions. (B) Floxed fzd7 mice (fzd7flox/flox) were generated. Homozygote floxed fzd7floxP/floxP mice and tie2-driven Cre-recombinase mice (Tie2-Cre) were crossed to generate fzd7 knockout mice specifically in the endothelium (fzd7ECKO). (C) Cre-recombinase expression in endothelial cells. Transgenic mice expressing Cre-recombinase driven by the Tie2 promoter were crossed with Rosa26-Stopfl-LacZ reporter mice. Staining of β-galactosidase (X-gal) in brain (b), muscle (c). EC-specific β-gal staining was detected only in Cre-Tie2 mice and in endothelial cells (d and e). (d) Immunostaining with anti-CD31 after X-gal staining in brain demonstrated the specificity in EC. (D) fzd7 mRNA expression quantified by quantitative RT–PCR in lung tissues from Wt and fzd7ECKO. fzd7 mRNA were normalized to GAPDH. (D) fzd7 mRNA expression quantified by quantitative RT–PCR in CD31(−) and CD31(+) cells from WT and fzd7ECKO mice. fzd7 mRNA were normalized to GAPDH.
To study the impact of fzd7 deletion on vascular integrity, fzd7ECKO and WT mice were evaluated for basal and induced vascular permeability. First, the leak of circulating Evan's blue dye in the skin was studied using the Miles assay in fzd7ECKO mice compared with wild-type (WT) littermate controls (Figure 3A). Based on vascular leakage measured by the Evans blue extravasation, fzd7ECKO mice displayed elevated vascular permeability at baseline compared with that in WT controls, which further increased under induction with VEGF (Figure 3A). Next, as inflammation is known to activate vascular permeability and oedema via different signalling pathways, we exposed mice to skin irritation and inflammation. Daily epicutaneous painting with 5% SDS, leads to an inflammatory response, marked by leucocyte invasion, epidermal hyperproliferation and oedema13 (Figure 3B, see Supplementary material online, Figure S2). After chronic cutaneous irritation, an increase in leucocyte infiltration was observed after 10 days both in WT and fzd7ECKO mice (see Supplementary material online, Figure S2A). The histological phenotype depicted in Figure 3B shows an increase in skin thickness and extensive oedema in fzd7ECKO compared with WT mice (Figure 3B).
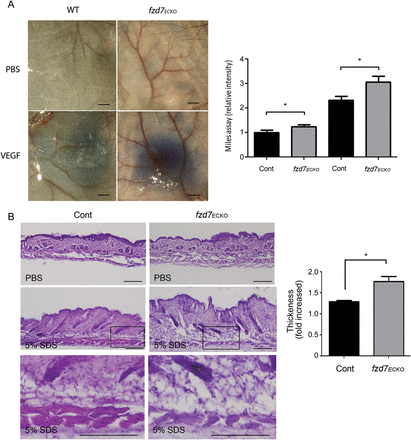
Fzd7 deletion increases vascular permeability in vivo. (A) Miles assay: representative picture of corresponding skin samples after local injection of PBS or VEGF for 1 h. Scale = 100 µm. Quantification by spectrophotometry (OD 620 nm), of extravased Evans blue in littermate, and fzd7ECKO mice under basal conditions (PBS) and after VEGF injection (VEGF). n = 8 mice/genotype from two independent experiments.*P < 0.05. (B) Chronic cutaneous inflammation model. Haematoxylin/eosin staining of skin after 10 days of SDS application in control and fzd7ECKO mice. Skin thickness induced by 5% SDS was evaluated as fold increase in skin thickening compared with PBS treatment in control mice (WT) vs. fzd7ECKO mice. Extensive oedema was observed at high magnification; black boxes identified the enlarged area. Scale 100 µm. n = 6−10 mice/genotype from two independent experiments. All the results are expressed as mean ± SEM. *P < 0.05.
Since Tie2-cre is not exclusively restricted to endothelium but is also active in haematopoietic cells and monocytes, we generated transgenic mice with fzd7 deletion in monocytes/macrophages and granulocyte (LysM-Cre/fzd7flox/flox) to rule out the role of leucocytes in the Tie2-Cre/fzd7flox/flox phenotype. Fzd7 deletion in myeloid cell lineage did not increase cutaneous basal permeability or modify VEGF-induced permeability (see Supplementary material online,Supplementary DataB). These data suggest that the phenotype observed in Tie2-Cre/fzd7flox/flox is due primarily to EC deletion.
All these results demonstrated that deletion of fzd7 in EC increased vascular permeability induced by different stimuli.
3.3 Loss of Fzd7 increases endothelial cell permeability and impairs endothelial cell–cell junction organization
The alteration of vascular permeability in fzd7ECKO mice compared with control WT littermates prompted us to analyse the functional role of Fzd7 on EC monolayer integrity and EC junction organization. We examined EC permeability after fzd7 depletion using two different human siRNAs on HUVEC (see Supplementary material online, Figure S3 and Dufourcq et al.8). Both control-siRNA and fzd7-siRNA transfected ECs grew to confluence to form a tight cell monolayer in Boyden chamber inserts. In unstimulated ECs, silencing of fzd7 induced a significant FITC-dextran accumulation in the lower chamber of the transwell compared with control-siRNA transfected EC (Figure 4A). VEGF stimulation rapidly promotes loss of cell–cell contacts in EC with an increase of FITC-dextran accumulation in the lower chamber. This leakage effect was significantly enhanced in fzd7-deficient EC monolayer (Figure 4A).
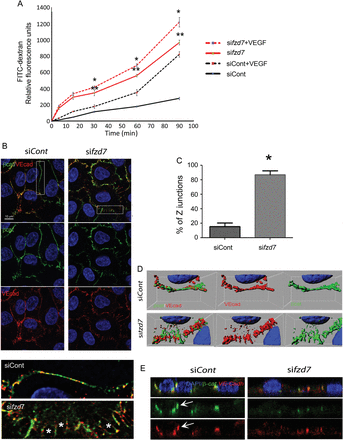
siRNA Fzd7 impairs paracellular permeability and cell junction organization. (A) Paracellular permeability quantified in vitro by FITC-dextran at baseline (line) and after exposure to VEGF (hashed line) for 5–90 min in siControl (black) and siFzd7 (red) treated cells. Results represent one of three independent experiments and are expressed as mean ± SD of triplicate experiments, **P < 0.001 siControl vs. sifzd7, *P < 0.005 siControl+VEGF vs. sifzd7+VEGF. (B) Confocal images of endothelial cell junctions after double staining with VE-cad (red) and β-cat (green) were performed after control siRNA (siCont) or anti-fzd7 siRNA (sifzd7) treatment. Representative high magnification image of endothelial cell junctions is presented. *Gaps between EC. (C) Quantification of jagged junction (Z junctions) in siControl- and siFzd7-treated cells,*P < 0.0001. (D) 3D image reconstruction and isosurface of β-catenin and VE-cadherin channels. (E) Analysis of confocal images in z-axis to study concentration of VE-cadherin/β-catenin complexes at points of cell–cell contact in control HUVEC and after sifzd7 treatment.
To determine whether the decreased paracellular permeability that was observed in fzd7 KD EC corresponded to disruption of endothelial adherence junction components, we examined VE-cadherin/ β-catenin expression patterns in confluent endothelial cell layers (Figure 4B–E). As examined by immunofluorescence microscopy, VE-cadherin and β-catenin co-localized and appeared in a linear distribution along cell–cell borders in control cells (Figure 4B). In contrast, KD of fzd7 expression induced a non-contiguous Z pattern for VE-cadherin/ β-catenin complexes with the opening of intercellular gaps in the monolayer (Figure 4B). Quantification of jagged junction revealed a strong increased of Z distribution of VE-cadherin/β-catenin staining in sifzd7 treated cells when compared with siRNA control (siControl)-treated cells, respectively, 87 ± 3 vs. 15 ± 2% of total junction (Figure 4C, P < 0.0001). Tri-dimensional confocal image reconstruction confirmed impairment of adherens junctions (Figure 4D). In sifzd7 conditions, z-axis analysis showed VE-cadherin and β-catenin diffusely expressed, and loss of focal localization in cell–cell junctions when compared with control conditions (Figure 4E).
In order to analyse TJ organization, Claudin5 and ZO-1 staining have been performed. Whereas no modification in Claudin5 expression was observed after sifzd7 treatment when compared with siControl treatment on HUVEC (see Supplementary material online, Figure S4), ZO-1 staining showed a more disrupted expression at the cell–cell junction after sifzd7 treatment compared with siControl-treated cells (see Supplementary material online,Supplementary Data). These results suggest that under fzd7 depletion, TJ are not well organized. These results demonstrated that Fzd7 plays a key role in the maintenance of endothelial cell integrity and promotes cell–cell junction assembly and organization.
3.4 Down-regulation of fzd7 impairs VE-cadherin and β-catenin expression and interaction at the cell membrane
We analysed the impact of fzd7 deletion on VE-cadherin and β-catenin expression in cultured cells and in mouse tissue. Immunoblot analysis on total cell lysats showed that the decrease of fzd7 expression led to a significant decrease in VE-cadherin and total β-catenin proteins (respectively, 58 ± 8 and 69 ± 10%, Figure 5A). β-catenin expression observed by immunofluorescence labelling showed a decrease in junctional expression around 30% in fzd7 KD-treated cells compared with siControl cells (Figure 5B). Using western blot, we analysed VE-cadherin and active β-catenin expression in total lung lysates from WT vs. fzd7ECKO mice. The results revealed a decrease in VE-cadherin and activated β-catenin expression in lung tissue from fzd7ECKO mice when compared with WT mice (n = 4, *P < 0.01) (Figure 5C). In vitro Fzd7 depletion decreased VE-cadherin/β-catenin association observed after immunoprecipitation and increased VE-cadherin and β-catenin tyrosine phosphorylation (Figure 5D).
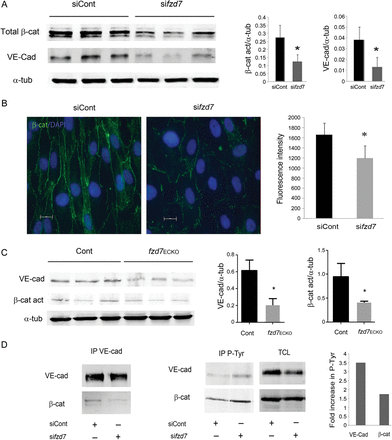
Fzd7 know-down or deletion decreases VE-cadherin and β-catenin expression. (A) WB analysis of VE-cadherin, total β-catenin, and α-tubulin expression in extracts of siControl- or sifzd7-treated HUVEC. Quantification of relative intensity was performed in triplicate and results are representative of four independent experiments (*P< 0.05). (B) A representative distribution of β-catenin analysed by immunofluorescence after siControl or sifzd7 treatment on HUVEC. Quantification of fluorescence intensity was measured in positive β-catenin junction. More than 100 junctions were measured for each condition. Mean values ± SEM, P< 0.001. (C) WB analysis of VE-cadherin, active β-catenin, and α-tubulin expression in lung lysates from WT vs. fzd7ECKO mice. Quantification of relative intensity was performed (n = 4, *P< 0.01). (D) Cell lysates immunoprecipitated with VE-Cadherin (left) or P-Tyr (right) and immunoblotted with either VE-cad or β-catenin. Total cell lysat (TCL) to detect VE-cadherin or β-catenin in siControl- and sifzd7-treated cells.
These results suggest that Fzd7 signalling regulates VE-cadherin/β-catenin expression at the EC junction and that Fzd7 may prevent VE-cadherin/β-catenin complex destabilization in confluent EC thereby contributing to the maintenance of cell–cell contacts in EC.
3.5 Down-regulation of fzd7 impairs canonical activation pathway
To better understand the molecular basis of Fzd7 function in endothelial cells, we investigated whether Fzd7 was able to regulate the canonical β-catenin-dependent pathway under basal condition and after Wnt activation. Using a TOP-Flash reporter construct for canonical β-catenin signalling (that contain TCF binding sites upstream of the luciferase transgene), we showed that fzd7 siRNA treatment decreased basal activity in EC when compared with siControl-treated cells (Figure 6A). Wnt3A CM and Wnt1 but not Wnt5A induced Top-Flash reporter activity in EC; this activation was significantly reduced in fzd7-depleted EC compared with control (Figure 6B). Analysis of canonical target gene expression by quantitative RT–PCR showed that fzd7 depletion in EC led to a decrease of both Axin2 and Lef1 transcripts (Figure 6C). Moreover, fzd7 siRNA led to a significant decrease of β-catenin at transcriptional levels (Figure 6D). These observations are consistent with the mode of signalling for the Fzd7 receptor, through a Wnt-canonical signalling.
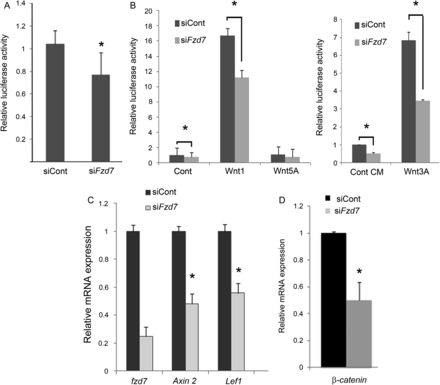
siRNA Fzd7 treatment decreases canonical Wnt signalling. (A) Luciferase reporter assay under basal conditions. HUVEC were co-transfected with a TOP-Flash construct including a luciferase reporter and control siRNA (siCont) or the reporter construct and Fzd7 siRNA (sifzd7). Luciferase activity was quantified after 48 h. Data from three or more representative experiments are represented as mean ± SD, *P < 0.05. (B) Luciferase reporter assay after Wnt activation. Control siRNA (siCont) or fzd7 siRNA (sifzd7) treated cells were co-cultured with CHO cells transfected with a plasmid harbouring β-gal (control), Wnt1, or Wnt5A or maintained with Wnt3A CM. Luciferase activity was quantified after 24 h. Data are represented as mean ± SD of triplicates from three representative experiments, *P< 0.005. (C) qRT-PCR analysis of Wnt-canonical target gene after siControl or sifzd7 treatment. The levels of mRNA were normalized to GAPDH. (D) Quantitative RT–PCR analysis of β-catenin expression on HUVEC treated with either siControl or sifzd7 and cultured for 48 h. The levels of mRNA were normalized to GAPDH. Data are represented as mean ± SD of triplicates from three representative experiments. *P< 0.05.
3.6 Activation of Wnt-canonical pathway rescues endothelial integrity and disruption of VE-cadherin-dependent EC junction under fzd7 depletion
LiCl has been shown to promote Wnt-canonical signalling by binding to and inactivating GSK-3β, thereby stabilizing β-catenin. To go further in the molecular mechanism by which Fzd7 controls EC permeability, LiCl was used to investigate whether GSK3 inhibition is able to rescue fzd7-KD-induced permeability and modification in VE-cadherin/β-catenin expression. Our data demonstrate that LiCl treatment prevented fzd7-KD induced permeability in HUVEC and MS1 (Figure 7A). Western blot on HUVEC demonstrated that LiCl treatment increased GSK-3β phosphorylation (Ser9), resulting in an increase in active β-catenin in both siControl- and sifzd7-treated cells (Figure 7B). Interestingly, LiCl totally prevented VE-cadherin down-regulation after fzd7 KD as shown in total cell lysates (Figure 7B). Using EC isolated from WT and fzd7ECKO mice, we demonstrated that in basal condition, primary fzd7ECKO EC presented a strong increase in paracellular permeability compared with WT EC (Figure 7C). Moreover, LiCl abolished endothelial permeability induced by fzd7 depletion in EC from fzd7ECKO mice (Figure 7C). We next investigated whether GSK3 inhibition is able to rescue VE-cadherin/β-catenin expression at the junction. Immunofluorescence analysis showed that LiCl induced a strong accumulation of junctional β-catenin in confluent EC from WT mice (Figure 7D). In accordance with data obtained in fzd7-KD HUVEC, β-catenin and VE-cadherin expression were impaired in fzd7ECKO EC compared with WT EC. In EC from fzd7ECKO mice, LiCl totally rescued β-catenin and VE-cadherin expression at the EC junction (Figure 7D).
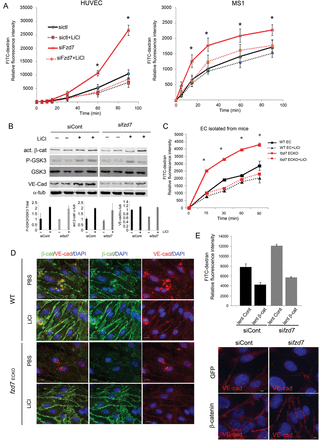
Endothelial integrity and β-catenin/VE-cadherin levels are maintained by activation of canonical Wnt pathway under fzd7 depletion. (A) HUVEC and MS1 cell transfected with siControl or sifzd7 were activated with LiCl for 24 h, or not, prior to measurement of the FITC-Dextran flux across the monolayer. Relative fluorescence was quantified after 5–90 min. Results are expressed as mean of FITC fluorescence intensity, *P < 0.001. (B) siRNA treated cells were activated or not with LiCl (30 mM) for 24 h. Cell lysates were immunoblotted with anti-VE-cadherin, total GSK3β or P-GSK3β (Ser9) or active β-catenin antibody. Representative WB of three independent experiments. (C) EC isolated from WT or fzd7ECKO mice were cultured on transwell and activated with LiCl for 24 h, or not, prior to measurement of the FITC-Dextran flux across the monolayer. Relative fluorescence was quantified after 15–90 min. *P < 0.001. (D) A representative distribution of β-catenin (green) and VE-cadherin (red) analysed by immunofluorescence after 24 h of LiCl or PBS treatment on EC from WT and fzd7ECKO mice. (E) siRNA-treated HUVEC were transduced with a lentiviral vector encoding eGFP (lent. Cont) or β-catenin (lent. β-cat). Two days after transduction, relative fluorescence of FITC-Dextran was quantified after 90 min to analyse endothelial permeability (n = 3). Junction organization was investigated after VE-cadherin staining in siRNA-treated cells after GFP or β-catenin overexpression.
To confirm the involvement of the Wnt-canonical pathway in Fzd7-induced signalling, EC permeability and VE-cadherin organization were investigated under conditions of β-catenin overexpression. Impairment of vascular permeability and VE-cadherin staining induced by fzd7 deletion were totally blocked by a lentiviral vector encoding β-catenin when compared with control GFP (Figure 7E).
These results suggest that Fzd7 initiates a GSK3β/β-catenin-dependent signalling cascade that is required for the regulation of VE-cadherin/β-catenin expression at the junction and for the functional integrity of the EC monolayer.
4. Discussion
Hyperpermeability triggered by inflammation or ischaemia causes oedema and tissue compression due to an increase in interstitial pressure and in turn may alter perfusion.3 Understanding the molecular mechanisms that regulate the properties of adherens junctions in EC will be important for the development of new therapeutic strategies to specifically modulate hyperpermeability in cardiovascular diseases, cancer, eyes, and inflammatory diseases. Several lines of evidence point to the role of Fzd receptors or Wnt/Fzd signalling in vascular formation during development or ischaemic diseases such as fzd4,14,22,23fzd5 genes,24 and sFRP-1.8,25 To date, the role of fzd7 in the vasculature has not been investigated. In the current report, we demonstrated for the first time that the Wnt receptor Fzd7 is a component of adherens junctions in the endothelium and that Wnt/Fzd pathway and adherens junctions are interconnected to regulate endothelium integrity. Fzd7 deletion strongly impaired EC permeability in vivo and in vitro, adherens junction organization, VE-cadherin/β-catenin expression as well as association at the junction. This study provides evidence of an autonomous endothelial cell-specific mechanism of cross-talk between Fzd7-induced Wnt-canonical signalling and VE-cadherin/ β-catenin expression.
We show that Fzd7 accumulates in points of cell–cell contact and co-localizes with VE-cadherin/β-catenin complexes. Immunoprecipitation experiments with different mutants suggest an interaction between Fzd7 and VE-cadherin through its intracellular domains. These reports are consistent with recent data showing that Fzd7 interacts with paraxial protocadherin (PAPC) or atypic Cadherin Flamingo to control cell–cell adhesion in Xenopus embryos.26 All these reports suggest that Fzd7 could participate in the persistence of cell–cell adhesion in the endothelium.
To address the role of fzd7 in the vasculature, we generated tissue deletion in EC using Tie2-Cre lox strategy. Fzd7ECKO mice are viable without gross alteration. These data are in agreement with the recent study showing that fzd7−/− mice are viable and fertile.27 We show that Fzd7 signalling is essential for vascular integrity and the control of vascular permeability. EC fzd7 deletion induced deregulation of different types of vascular permeability: (i) basal vascular permeability of normal tissues; (ii) acute vascular hyper-permeability that occurs in response to VEGF-A; and (iii) chronic vascular hyperpermeability that characterizes pathological angiogenesis. Impairment of endothelium integrity correlated with the increase in paracellular permeability in vitro observed after fzd7 deletion. Our results demonstrated that Fzd7 signalling played a crucial role in adult vasculature by controlling endothelial permeability.
Endothelial permeability is regulated in part by the dynamic organization of β-catenin/VE-cadherin complexes. Change in the structure and composition of adherens junctions may have profound effects on vascular permeability as well as on the overall vascular homeostasis.28 The role of fzd7 on cadherin expression regulation has been described in cancer progression. RNAi-mediated depletion of fzd7 resulted in the persistence of a mesenchymal state, which was paralleled by decreased E-cadherin expression and cell proliferation.29 In vascular biology, modification of VE-cadherin expression or function alters vascular morphogenesis in the embryo and vascular permeability in the adult. In vivo disruption of VE-cadherin with a function-blocking antibody leads to vascular leak and haemorrhage.30 Partial reduction of VE-cadherin expression in zebrafish embryo affects vascular integrity by inducing vascular instability and haemorrhages.31 Here we report the first evidence of direct functional cross-talk between a Frizzled receptor and adherens junction components in vascular cells. fzd7 depletion induced impairment of junction organization with the occurrence of gaps between EC and a decrease of VE-cadherin and β-catenin expression at the cell–cell junction. Moreover, loss of fzd7 induces changes in tyrosine phosphorylation of both cadherins and catenins which is associated with weak junctions, junction dissociation, and impairment of barrier function.1 In this study, we mostly focus on the role of Fzd7/β-catenin signalling on adherens junction organization and permeability control, however, other barrier-associated protein may also be controlled by Fzd7 signalling. Many reports have suggested that AJs and TJs are interconnected and that AJs influence TJ organization. However, the signals transferred by AJs that promote TJ assembly are still largely unknown.32,33 While no modification in Claudin5 TJ protein was observed after fzd7 depletion, ZO-1 distribution was found to be impaired in HUVEC depleted of fzd7. ZO-1 can associate with different TJ proteins such as claudins, occludin, or junctional adhesion molecules,34 suggesting that fzd7 depletion could also impair TJs organization.
Fzd7 orchestrates different processes by activating both canonical and non-canonical Wnt signalling depending on the cell and tissue context.35 Here, we present evidence that Fzd7 mediates the Wnt-canonical pathway in EC culture assays. In EC, decreased fzd7 expression resulted in a decrease in the β-catenin transactivation activities in basal conditions and after Wnt1 and Wnt3A activation associated with a decrease in β-catenin target gene expression. Moreover, activation of the Wnt-canonical pathway by LiCl or β-catenin overexpression rescued fzd7 KD-induced permeability, prevented the decrease of VE-cadherin/β-catenin expression, and adherens junction disorganization observed after fzd7 down-regulation or depletion. These results demonstrated that in the endothelium Fzd7 participates through the Wnt-canonical pathway in the control of endothelial permeability. Reduction in the amount of β-catenin protein could be explained by insufficiency of Frizzled signalling, which then leads to an overly active β-catenin destruction complex. Moreover, loss of fzd7 expression decreased β-catenin gene (Ctnnb1) transcription, since mRNA expression was reduced under fzd7 KD. These results suggested that Fzd7, in regulating β-catenin intracellular levels, affects β-catenin dual transactivating activity and junctional function. A central role for canonical Wnt signalling in CNS vascular development has independently emerged from studies of mice carrying targeted mutations in the genes coding for Wnt7a, Wnt7b, and β-catenin.36–38 Moreover, EC-specific deletion or gain-of-function of β-catenin, alters development of the murine embryonic vasculature and results in early lethality in utero, with changes in the vascular lumen and defects of vascular remodelling.36,39 While the complete mechanism of vascular permeability regulation has not yet been fully elucidated, our results are consistent with the fact that Wnt/Fzd7/β-catenin signalling appears to be necessary to control vascular permeability and maintain vascular integrity in adults and in non-CNS blood vessels.
In conclusion, these data contribute to a better understanding of the complexity of the Wnt/Fzd pathway, underlining the fine tuning and functional specificity of the Fzd receptor in the endothelium. This study demonstrates how Fzd7 regulates paracellular permeability via the canonical Wnt pathway and a direct functional cross-talk with VE-cadherin-mediated cell–cell junctions. This new Wnt/Fzd7 pathway could help in the development of new therapeutic strategies to specifically modulate excess permeability in ischaemic diseases, chronic inflammation, or cancer.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by grants from the Agence Nationale de la Recherche (ANR-2010-Blanc-1135-01), from Region Aquitaine (20101301039MP), and from Communauté de Travail Pyrénéenne. N.F.T. was supported by a gift from Pr. F. Fontan.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}